Chemosensitivity of Patient-Derived Cancer Stem Cells Identifies Colorectal Cancer Patients with Potential Benefit from FGFR Inhibitor Therapy

 , ,
, , 
Abstract
:1. Introduction
2. Results
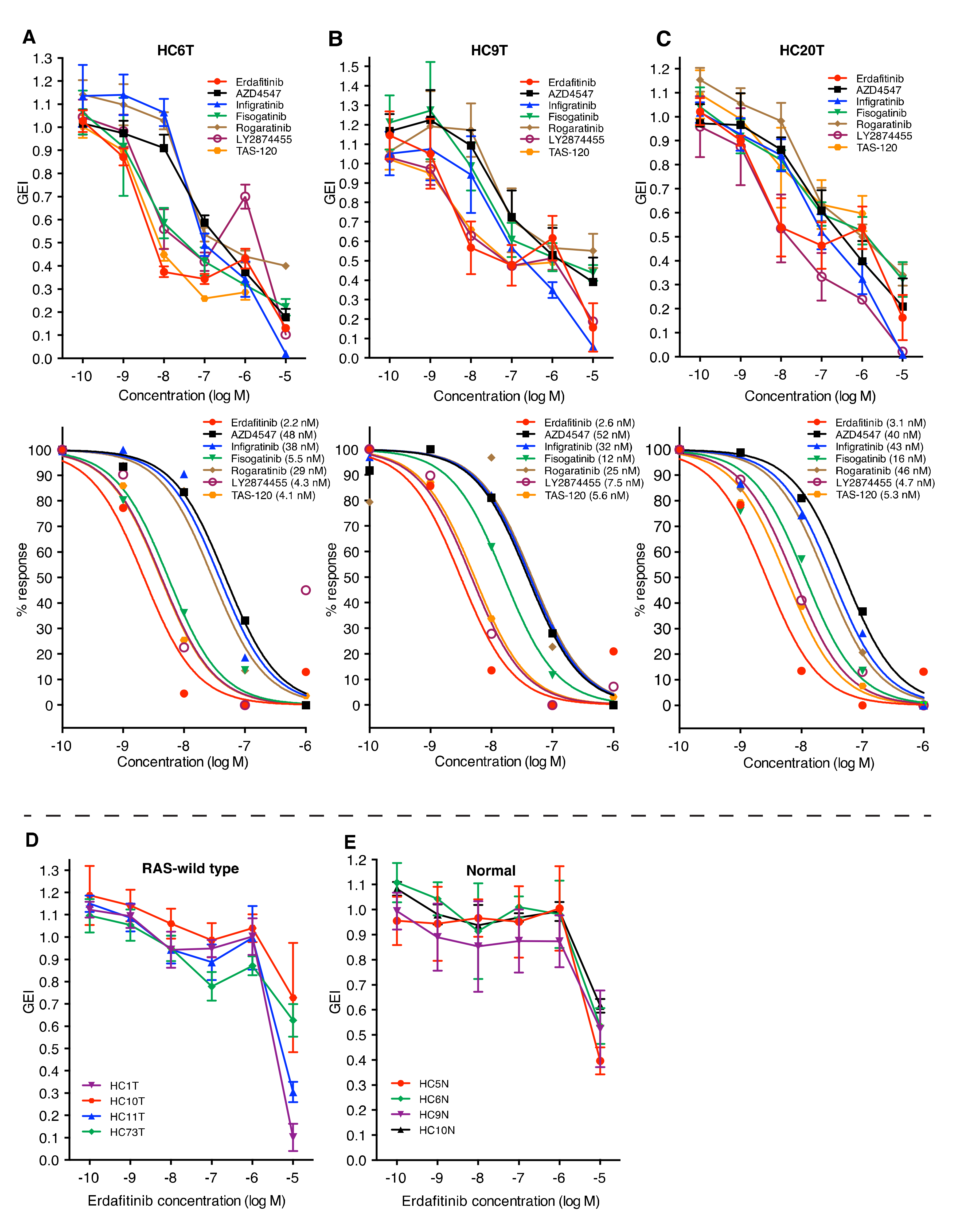
2.1. The in vitro Evaluation of the FGFR Inhibitors for Colorectal Cancer Pateints by Using Patient-Derived TIC Spheroids
2.2. FGFR Inhibitors Suppress in vitro Growth of Colorectal Cancer TIC Spheroids Derived from a Subset of Patients
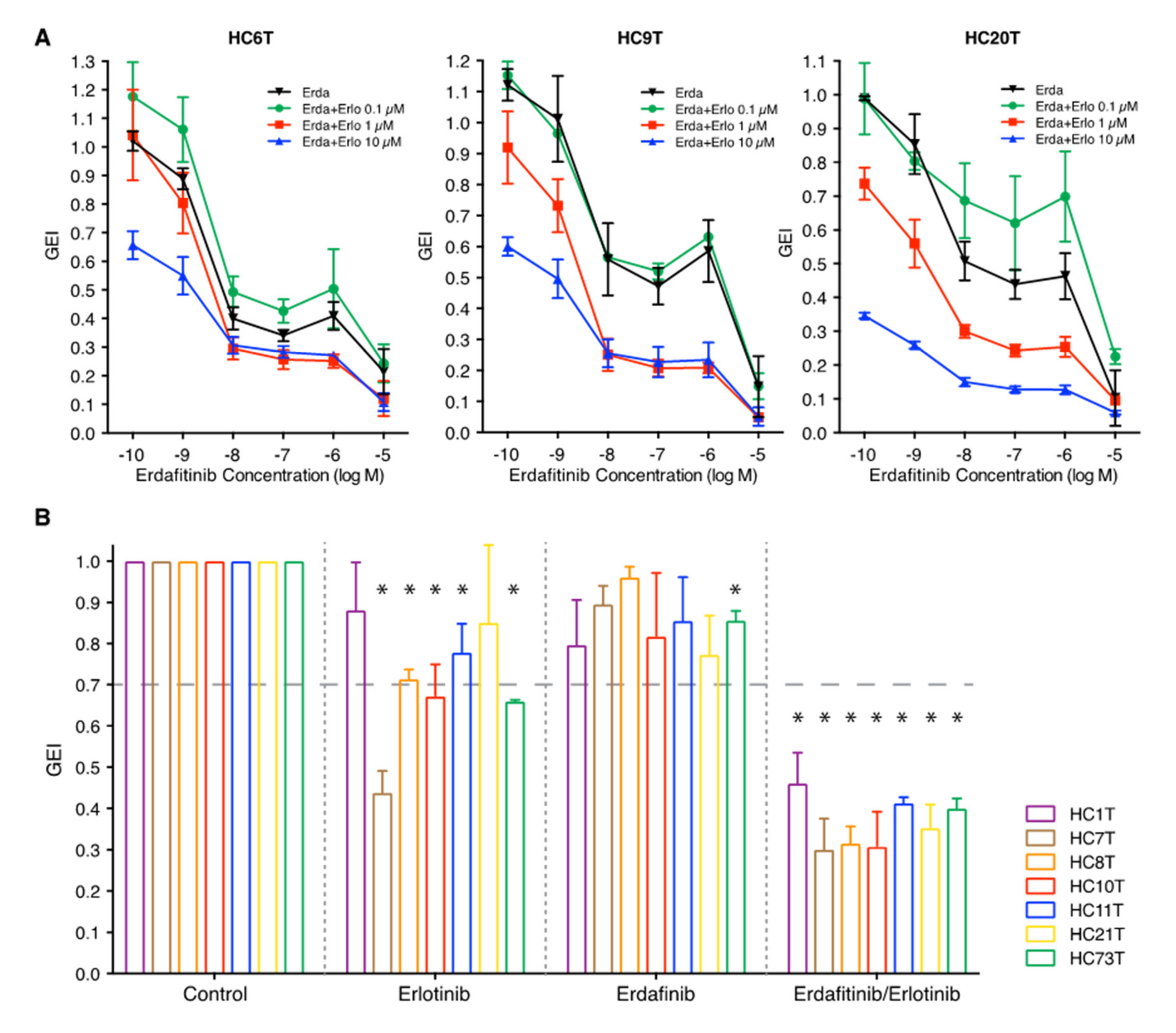
2.3. Synergistic in vitro Growth Suppression by Combination Treatment with FGFR/EGFR Inhibitors
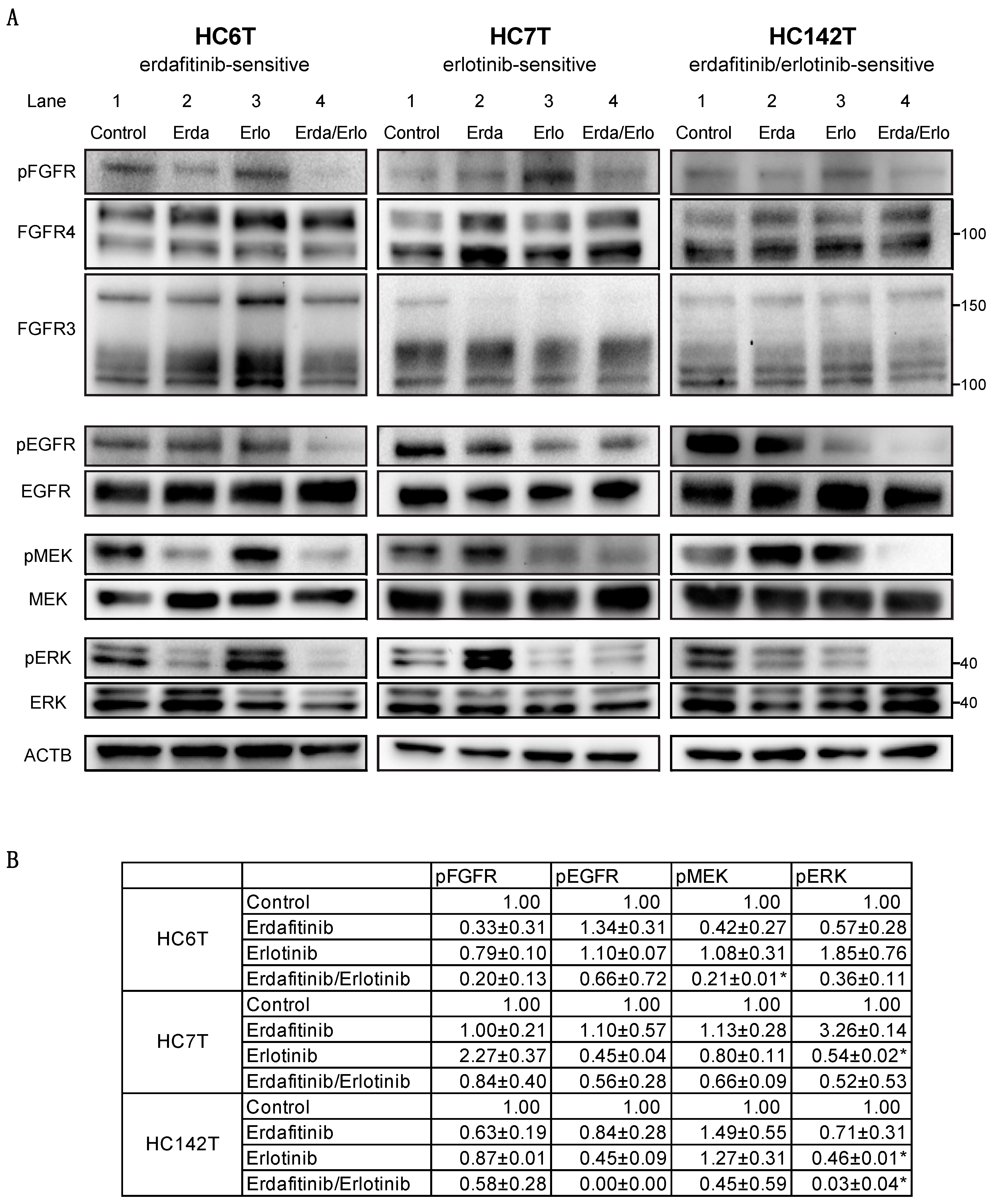
2.4. Immunoblotting Analysis of Colorectal Cancer Spheroid Lines for FGFR, EGFR, and Downstream Effector Proteins
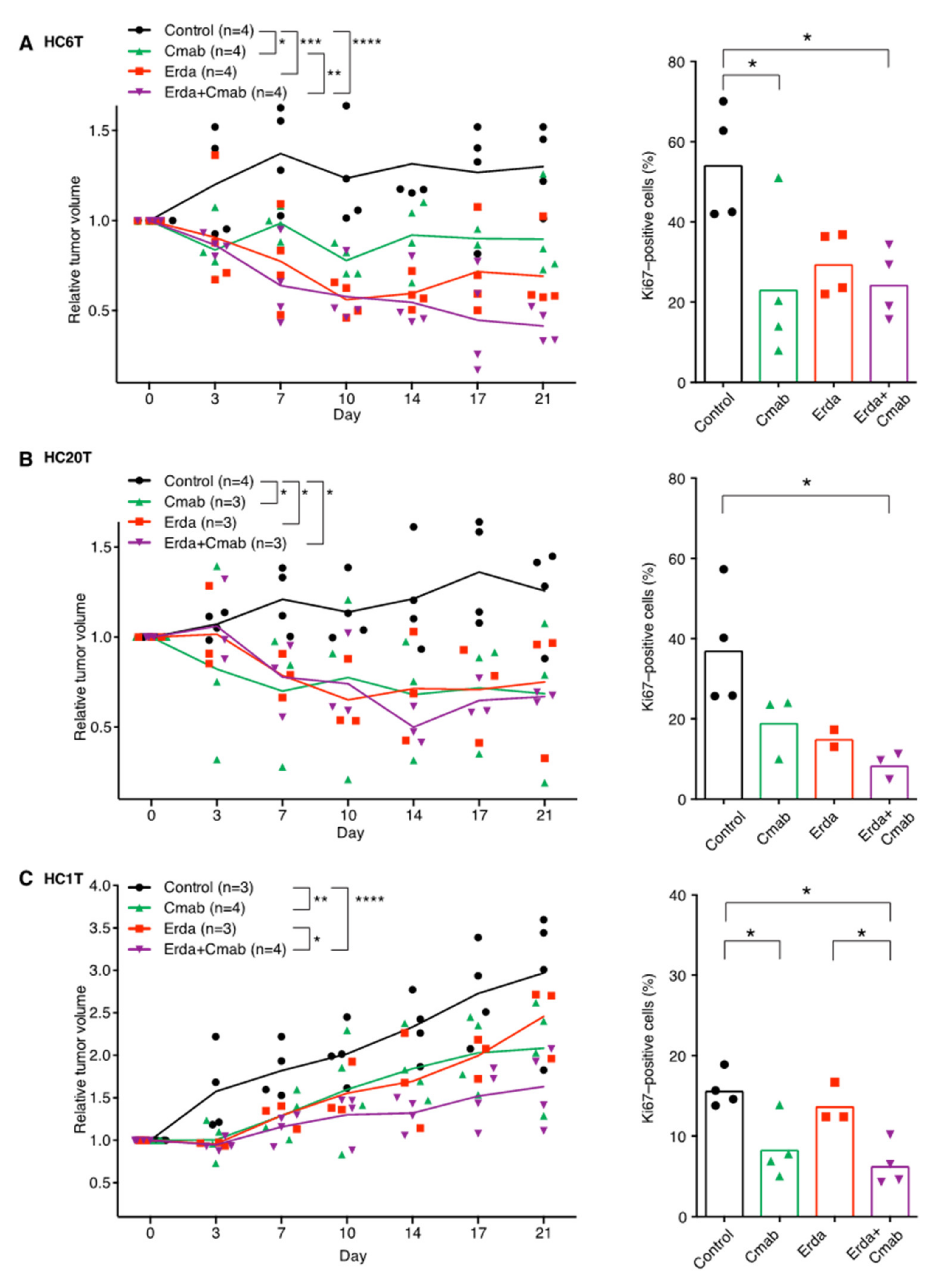
2.5. Erdafitinib and/or Cetuximab Reduce the Size of Patient-Derived Spheroid-Xenograft (PDSX) Tumors at Clinically Relevant Doses
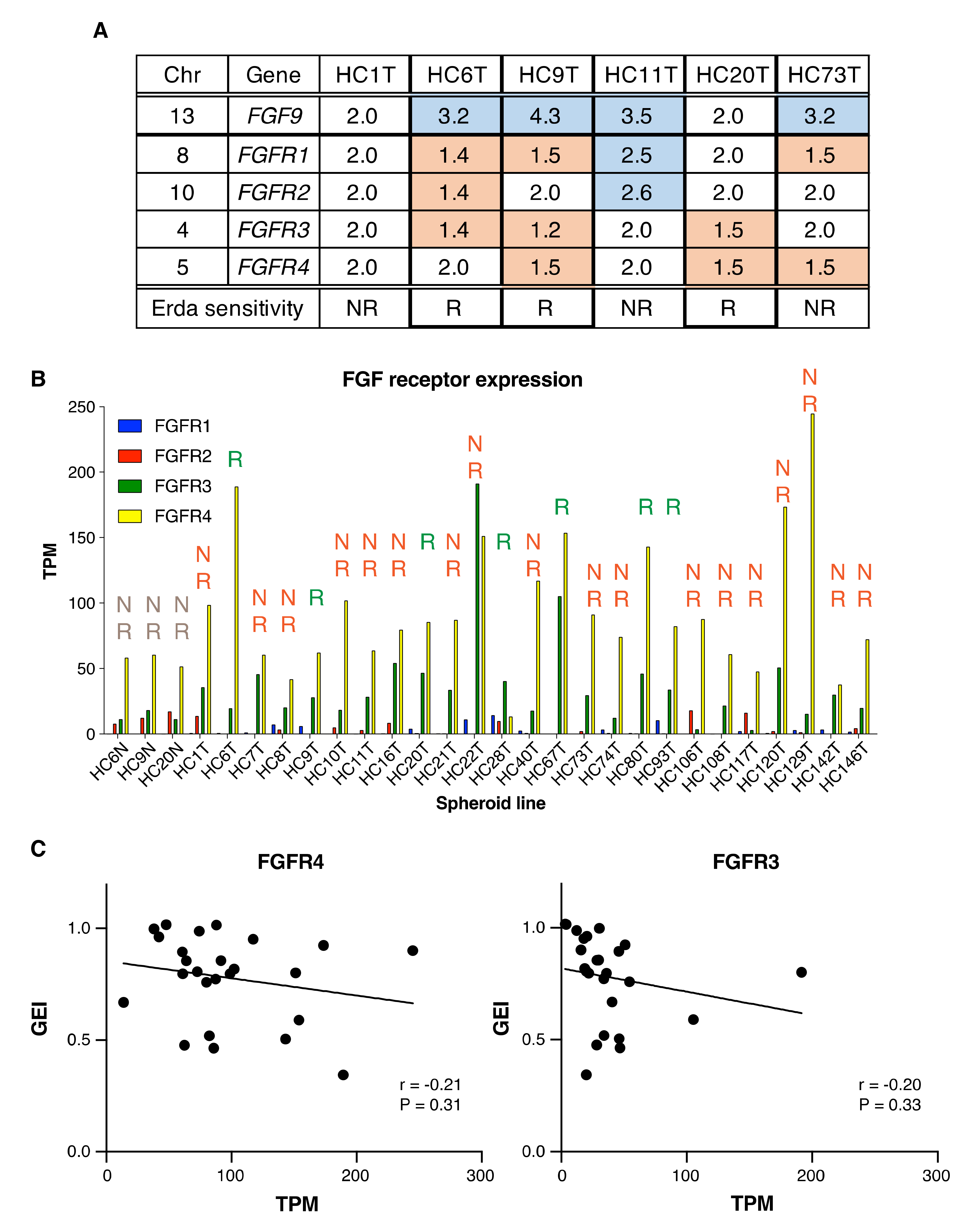
2.6. Little Association between FGFR Inhibitor Sensitivity and FGFR Gene Alterations, or mRNA Levels in Colorectal Cancer Spheroids
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Patient-Derived Cancer-Spheroid Culture
4.3. Immunoblotting Analysis
4.4. Chemicals
4.5. Luminescence-Based Drug-Dosing Tests in Spheroid Culture
4.6. Preparation of Spheroid DNA and RNA Samples
4.7. Array-Based Comparative Genomic Hybridization (CGH) Analysis
4.8. Mutation Analysis
4.9. Filtration of Variants
4.10. RNA-Seq Analysis
4.11. PDSX (Patient-Derived Spheroid Xenograft)
4.12. Drug Sensitivity Tests in Mice
4.13. Histological Examinations
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araujo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, L.; Su, X.; Li, M.; Xie, L.; Malchers, F.; Fan, S.; Yin, X.; Xu, Y.; Liu, K.; et al. Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clin. Cancer Res. 2012, 18, 6658–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Bang, Y.J.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.R.; Smith, N.R.; Frewer, P.; Ratnayake, J.; et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann. Oncol. 2017, 28, 1316–1324. [Google Scholar] [CrossRef]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A phase Ib open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [Green Version]
- Saka, H.; Kitagawa, C.; Kogure, Y.; Takahashi, Y.; Fujikawa, K.; Sagawa, T.; Iwasa, S.; Takahashi, N.; Fukao, T.; Tchinou, C.; et al. Safety, tolerability and pharmacokinetics of the fibroblast growth factor receptor inhibitor AZD4547 in Japanese patients with advanced solid tumours: A Phase I study. Invest. New Drugs. 2017, 35, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, T.; Takahashi, S.; Iwasawa, R.; Noguchi, H.; Aoki, M.; Doi, T. Safety, pharmacokinetic, and pharmacodynamics of erdafitinib, a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients with advanced or refractory solid tumors. Invest. New Drugs. 2018, 36, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase I, dose-escalation and dose-expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [PubMed]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.P.; et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1-3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1454–1466. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Maekawa, H.; Kakizaki, F.; Yamaura, T.; Kawada, K.; Sakai, Y.; Taketo, M.M. An improved method for culturing patient-derived colorectal cancer spheroids. Oncotarget 2018, 9, 21950–21964. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, H.; Miyoshi, H.; Yamaura, T.; Itatani, Y.; Kawada, K.; Sakai, Y.; Taketo, M.M. A chemosensitivity study of colorectal cancer using xenografts of patient-derived tumor initiating cells. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, S.; Politz, O.; Bender, S.; Heroult, M.; Lustig, K.; Thuss, U.; Kneip, C.; Kopitz, C.; Zopf, D.; Collin, M.P.; et al. Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer. 2019, 145, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef]
- US Food and Drug Administration FDA Approves Erbitux for Colorectal Cancer. Available online: www.Fda.Gov (accessed on 19 July 2019).
- US Food and Drug Administration FDA Approves Vectibix (Panitumumab) to Treat Metastatic Colorectal Carcinoma. Available online: www.Fda.Gov (accessed on 19 July 2019).
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Tournigand, C.; Chibaudel, B.; Samson, B.; Scheithauer, W.; Vernerey, D.; Mesange, P.; Lledo, G.; Viret, F.; Ramee, J.F.; Tubiana-Mathieu, N.; et al. Bevacizumab with or without erlotinib as maintenance therapy in patients with metastatic colorectal cancer (GERCOR DREAM; OPTIMOX3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 1493–1505. [Google Scholar] [CrossRef]
- Heuertz, S.; Le Merrer, M.; Zabel, B.; Wright, M.; Legeai-Mallet, L.; Cormier-Daire, V.; Gibbs, L.; Bonaventure, J. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur. J. Hum. Genet. 2006, 14, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Faltas, B.M. The molecular limitations of biomarker research in bladder cancer. World J. Urol. 2019, 37, 837–848. [Google Scholar] [CrossRef]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: Relevance to cancer therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar] [CrossRef]
- Triantis, V.; Saeland, E.; Bijl, N.; Oude-Elferink, R.P.; Jansen, P.L. Glycosylation of fibroblast growth factor receptor 4 is a key regulator of fibroblast growth factor 19-mediated down-regulation of cytochrome P450 7A1. Hepatology 2010, 52, 656–666. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Silterra, J.; Schwartz, S.; Wong, B.; Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P.; Airoldi, E.M.; Burge, C.B. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 2015, 31, 2400–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontier, D.; Picart, C.; El Baidouri, M.; Roudier, F.; Xu, T.; Lahmy, S.; Llauro, C.; Azevedo, J.; Laudie, M.; Attina, A.; et al. The m(6)A pathway protects the transcriptome integrity by restricting RNA chimera formation in plants. Life Sci. Alliance 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Muller, S.; Gartner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002, 62, 840–847. [Google Scholar] [PubMed]
- Frullanti, E.; Berking, C.; Harbeck, N.; Jezequel, P.; Haugen, A.; Mawrin, C.; Parise, O., Jr.; Sasaki, H.; Tsuchiya, N.; Dragani, T.A. Meta and pooled analyses of FGFR4 Gly388Arg polymorphism as a cancer prognostic factor. Eur. J. Cancer Prev. 2011, 20, 340–347. [Google Scholar] [CrossRef]
- US Food and Drug Administration FDA Approves Balversa for Colorectal Cancer. Available online: www.Fda.Gov (accessed on 19 July 2019).
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, H.; Yun, M.R.; Kang, H.N.; Pyo, K.H.; Park, H.J.; Lee, J.M.; Choi, H.M.; Ellinghaus, P.; Ocker, M.; et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis 2016, 5, e241. [Google Scholar] [CrossRef] [Green Version]
- Hatlen, M.A.; Schmidt-Kittler, O.; Sherwin, C.A.; Rozsahegyi, E.; Rubin, N.; Sheets, M.P.; Kim, J.L.; Miduturu, C.; Bifulco, N.; Brooijmans, N.; et al. Acquired on-target clinical resistance validates FGFR4 as a driver of hepatocellular carcinoma. Cancer Discov. 2019, 9, 1686–1695. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013, 3, 1058–1071. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer. 2019, 19, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef]
- Maughan, T.S.; Adams, R.A.; Smith, C.G.; Meade, A.M.; Seymour, M.T.; Wilson, R.H.; Idziaszczyk, S.; Harris, R.; Fisher, D.; Kenny, S.L.; et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 2011, 377, 2103–2114. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; VanDussen, K.L.; Malvin, N.P.; Ryu, S.H.; Wang, Y.; Sonnek, N.M.; Lai, C.W.; Stappenbeck, T.S. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 2017, 36, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Miyake, N.; Yoshimura, J.; Okamura, K.; Niihori, T.; Saitsu, H.; Doi, K.; Shimizu, M.; Nakabayashi, K.; Aoki, Y.; et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 2016, 61, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, M.; Yasuda, J.; Katsuoka, F.; Nariai, N.; Kojima, K.; Kawai, Y.; Yamaguchi-Kabata, Y.; Yokozawa, J.; Danjoh, I.; Saito, S.; et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 2015, 6, 8018. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.R.; Yang, Z.; Dong, H.; Camuso, A.; McGlinchey, K.; Fager, K.; Flefleh, C.; Kan, D.; Inigo, I.; Castaneda, S.; et al. Correlation of pharmacokinetics with the antitumor activity of Cetuximab in nude mice bearing the GEO human colon carcinoma xenograft. Cancer Chemother. Pharmacol. 2005, 56, 455–464. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Patient | Tumor | Pathological Stage | Mutations in Representative Cancer-Related Genes | Drug Sensitivity c (GEI) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | Sex | Location a | Histology b | Erdafitinib | Erlotinib | Erdafitinib/Erlotinib | |||||||||
| (FGFR Inhibitor) | (EGFR Inhibitor) | Combination | |||||||||||||
| HC6T | 57 | M | S | Well-Mod | 4 | APC | TP53 | R (0.34) | NR | R (0.25) | |||||
| HC9T | 73 | M | A | Well-Mod | 2 | APC | TP53 | R (0.47) | NR | R (0.20) | |||||
| HC20T | 66 | F | S | Well-Mod | 4 | TP53, | R (0.43) | NR | R (0.24) | ||||||
| HC28T | 71 | M | D | Well-Mod | 2 | APC | PIK3CA | R (0.66) | NR | R (0.25) | |||||
| HC67T | 72 | M | R | Well-Mod | 2 | APC | R (0.58) | NR | R (0.28) | ||||||
| HC80T | 75 | M | S | Well-Mod | 4 | APC | TP53 | R (0.50) | NR | R (0.32) | |||||
| HC93T | 66 | F | R | Well-Mod | 3 | TP53 | R (0.51) | NR | R (0.28) | ||||||
| HC7T | 68 | F | A | Well-Mod | 2 | TP53 | NR | R (0.43) | R (0.30) | ||||||
| HC10T | 70 | F | C | Well-Mod | 2 | APC | TP53 | NR | R (0.67) | R (0.30) | |||||
| HC73T | 75 | M | S | Well-Mod | 4 | APC | TP53 | NR | R (0.65) | R (0.39) | |||||
| HC108T | 42 | F | R | Well-Mod | 4 | APC | TP53 | NR | R (0.57) | R (0.31) | |||||
| HC117T | 77 | M | S | Mucinous | 3 | APC | TP53 | NR | R (0.62) | R (0.20) | |||||
| HC1T | 81 | F | A | Well-Mod | 4 | APC | PIK3CA | NR | NR | R (0.46) | |||||
| HC8T | 66 | F | T | Well-Mod | 1 | APC | PIK3CA | FBXW7 | NR | NR | R (0.31) | ||||
| HC11T | 74 | M | T | Mucinous | 3 | APC | TP53 | NR | NR | R (0.41) | |||||
| HC16T | 89 | M | R | Well-Mod | 3 | APC | TP53 | FBXW7 | NR | NR | R (0.57) | ||||
| HC21T | 52 | M | R | Well-Mod | 4 | APC | TP53 | FBXW7 | NR | NR | R (0.35) | ||||
| HC22T | 51 | M | S | Well-Mod | 2 | APC | TP53 | NR | NR | R (0.58) | |||||
| HC74T | 50 | M | S | Well-Mod | 4 | APC | TP53 | NR | NR | R (0.67) | |||||
| HC142T | 67 | F | R | Well-Mod | 4 | NR | NR | R (0.34) | |||||||
| HC146T | 45 | F | R | Well-Mod | 4 | TP53 | NR | NR | R (0.36) | ||||||
| HC40T | 82 | M | T | Well-Mod | 3 | APC | TP53 | PIK3CA | NR | NR | NR | ||||
| HC106T | 63 | F | T | Well-Mod | 1 | APC | NR | NR | NR | ||||||
| HC120T | 55 | M | S | Well-Mod | 3 | TP53 | NR | NR | NR | ||||||
| HC129T | 55 | M | R | Well-Mod | 2 | APC | TP53 | NR | NR | NR | |||||
| Sensitivity | 7 (28%) | 5 (20%) | 21 (84%) | ||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, T.; Miyoshi, H.; Kakizaki, F.; Maekawa, H.; Yamaura, T.; Morimoto, T.; Katayama, T.; Kawada, K.; Sakai, Y.; Taketo, M.M. Chemosensitivity of Patient-Derived Cancer Stem Cells Identifies Colorectal Cancer Patients with Potential Benefit from FGFR Inhibitor Therapy. Cancers 2020, 12, 2010. https://doi.org/10.3390/cancers12082010
Yamamoto T, Miyoshi H, Kakizaki F, Maekawa H, Yamaura T, Morimoto T, Katayama T, Kawada K, Sakai Y, Taketo MM. Chemosensitivity of Patient-Derived Cancer Stem Cells Identifies Colorectal Cancer Patients with Potential Benefit from FGFR Inhibitor Therapy. Cancers. 2020; 12(8):2010. https://doi.org/10.3390/cancers12082010
Chicago/Turabian StyleYamamoto, Takehito, Hiroyuki Miyoshi, Fumihiko Kakizaki, Hisatsugu Maekawa, Tadayoshi Yamaura, Tomonori Morimoto, Toshiro Katayama, Kenji Kawada, Yoshiharu Sakai, and M. Mark Taketo. 2020. "Chemosensitivity of Patient-Derived Cancer Stem Cells Identifies Colorectal Cancer Patients with Potential Benefit from FGFR Inhibitor Therapy" Cancers 12, no. 8: 2010. https://doi.org/10.3390/cancers12082010