The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies

Abstract
:1. Introduction
2. Literature Search Strategy
3. The CRC-Associated Pro-Inflammatory Milieu
3.1. Cellular Components of the CRC Microenvironment
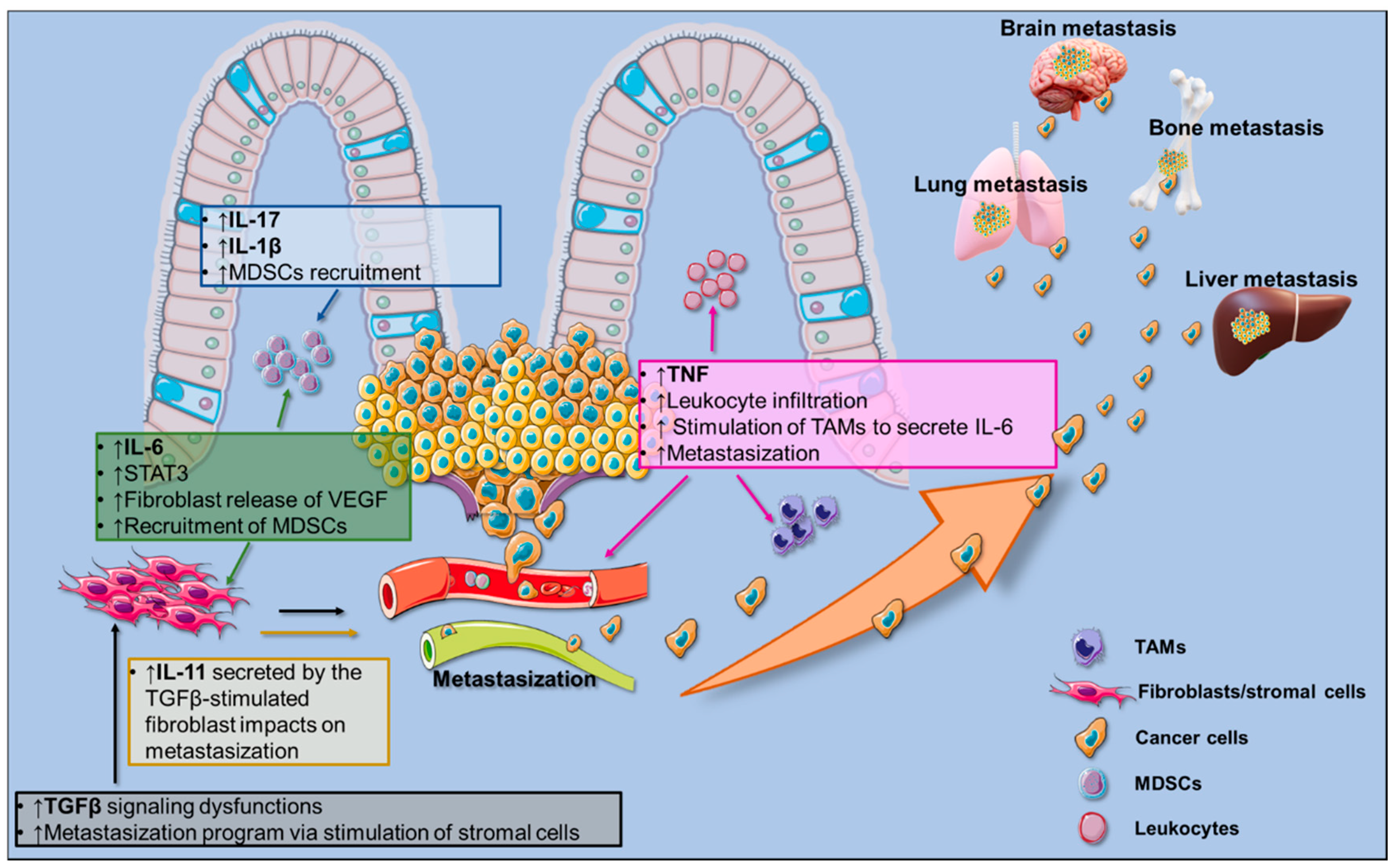
3.2. Pro-Inflammatory Signals in CRC
4. Pro-Resolving Lipid Mediators: A Brief Overview
5. The Resolution of CRC-Associated Inflammation through the ω-3 PUFAs: A Lesson from Clinical and Animal Studies
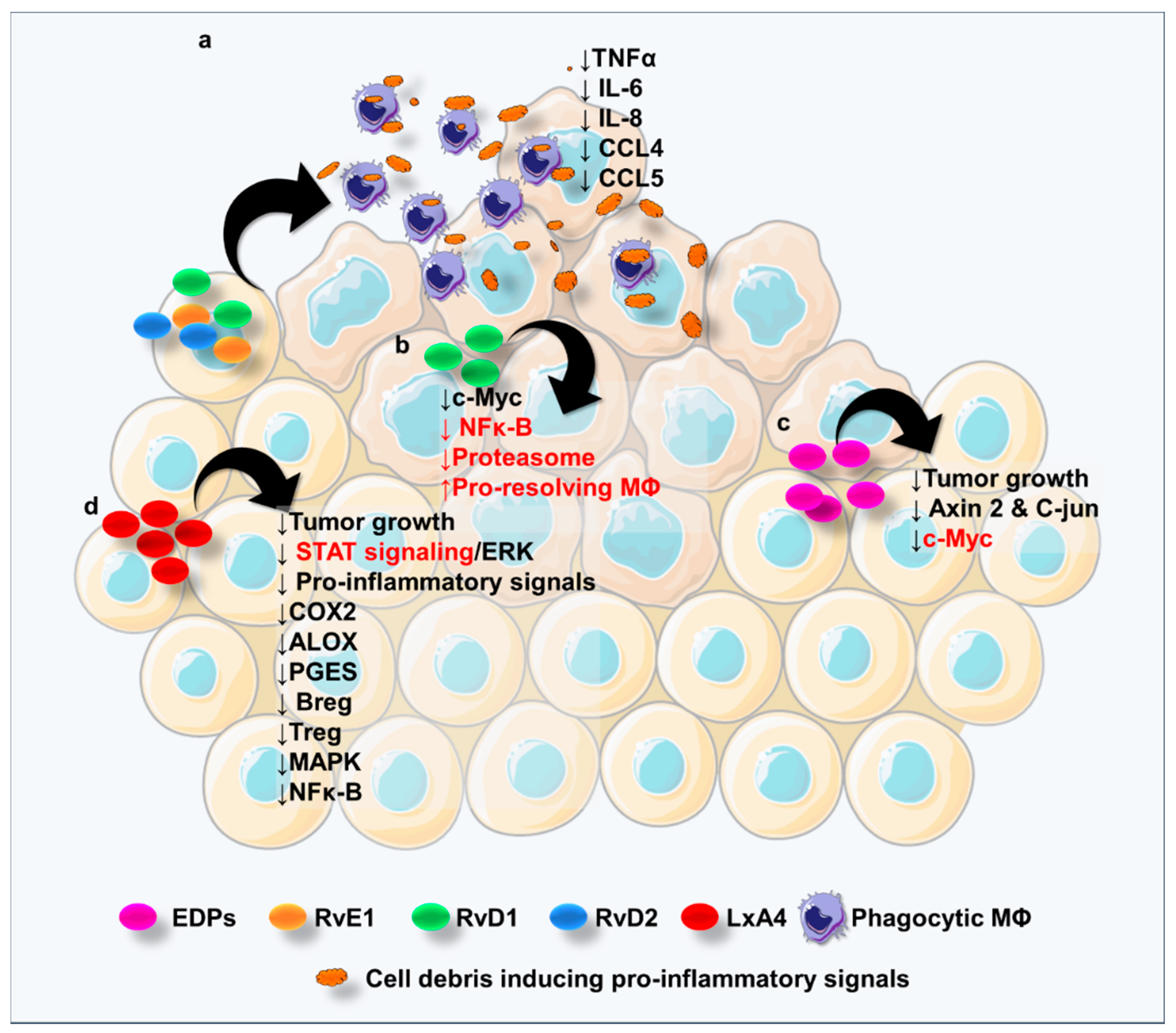
6. Specialized Pro-Resolving Lipid Mediators in Colorectal Cancer
6.1. SPMs Derived from ALA
6.2. SPMs Derived from LA
7. PUFA Receptor-Mediated Signaling in CRC
8. Pharmacology and Diet as a Possible Approach to Enhance the Lipid-Mediated CRC-Associated Resolution of Inflammation
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Valderrama-Treviño, A.I.; Barrera-Mera, B.; Ceballos-Villalva, J.C.; Montalvo-Javé, E.E. Hepatic Metastasis from Colorectal Cancer. Euroasian J. Hepato-Gastroenterol. 2017, 7, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Hu, J.; Yang, D.; Cosgrove, D.P.; Xu, R. Pattern of distant metastases in colorectal cancer: A SEER based study. Oncotarget 2015, 6, 38658–38666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal. Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Van Blarigan, E.L.; Fuchs, C.S.; Niedzwiecki, D.; Zhang, S.; Saltz, L.B.; Mayer, R.J.; Mowat, R.B.; Whittom, R.; Hantel, A.; Benson, A.; et al. Association of Survival With Adherence to the American Cancer Society Nutrition and Physical Activity Guidelines for Cancer Survivors After Colon Cancer Diagnosis. JAMA Oncol. 2018, 4, 783. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef]
- Nelson, N. On Trial: Evidence From Using Aspirin to Prevent Cancer. J. Natl. Cancer Inst. 2015, 107, 265. [Google Scholar] [CrossRef] [Green Version]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Suh, O.; Mettlin, C.; Petrelli, N.J. Aspirin use, cancer, and polyps of the large bowel. Cancer 1993, 72, 1171–1177. [Google Scholar] [CrossRef]
- Mohammed, A.; Yarla, N.S.; Madka, V.; Rao, C.V. Clinically Relevant Anti-Inflammatory Agents for Chemoprevention of Colorectal Cancer: New Perspectives. Int. J. Mol. Sci. 2018, 19, 2332. [Google Scholar] [CrossRef] [Green Version]
- Suh, N.; Reddy, B.S.; DeCastro, A.; Paul, S.; Lee, H.J.; Smolarek, A.K.; So, J.Y.; Simi, B.; Wang, C.X.; Janakiram, N.B.; et al. Combination of Atorvastatin with Sulindac or Naproxen Profoundly Inhibits Colonic Adenocarcinomas by Suppressing the p65/-Catenin/Cyclin D1 Signaling Pathway in Rats. Cancer Prev. Res. 2011, 4, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins. Leukot. Essent. Fatty Acids 2005, 73, 141–162. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, B.; Li, Y. Resolution of Cancer-Promoting Inflammation: A New Approach for Anticancer Therapy. Front. Immunol. 2017, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef]
- Ungaro, F.; Rubbino, F.; Danese, S.; D’Alessio, S. Actors and Factors in the Resolution of Intestinal Inflammation: Lipid Mediators As a New Approach to Therapy in Inflammatory Bowel Diseases. Front. Immunol. 2017, 8, 1331. [Google Scholar] [CrossRef]
- Ungaro, F.; Tacconi, C.; Massimino, L.; Corsetto, P.A.; Correale, C.; Fonteyne, P.; Piontini, A.; Garzarelli, V.; Calcaterra, F.; Della Bella, S.; et al. MFSD2A Promotes Endothelial Generation of Inflammation-resolving Lipid Mediators and Reduces Colitis in Mice. Gastroenterology 2017, 153, 924. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Langmead, C.J.; Riddy, D.M. New Advances in Targeting the Resolution of Inflammation: Implications for Specialized Pro-Resolving Mediator GPCR Drug Discovery. ACS Pharmacol. Transl. Sci. 2020, 3, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, M.K.; Lee, E.J.; Lee, C.H. Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int. J. Biochem. Cell Biol. 2013, 45, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Marginean, A.; Sharma-Walia, N. Lipoxins exert antiangiogenic and anti-inflammatory effects on Kaposi’s sarcoma cells. Transl. Res. 2015, 166, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.M.; Neto, E.H.; Figueiredo, C.C.; Barja Fidalgo, C.; Fierro, I.M.; Morandi, V. ATL-1, a synthetic analog of lipoxin, modulates endothelial permeability and interaction with tumor cells through a VEGF-dependent mechanism. Biochem. Pharmacol. 2014, 90, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Serhan, C.N. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br. J. Pharmacol. 2019, 176, 1024–1037. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Janakiram, N.B.; Rao, C.V. The Role of Inflammation in Colon Cancer. Adv. Exp. Med. Biol. 2014, 816, 25–52. [Google Scholar]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.K.; Rybicki, L.A.; Goldblum, J.R.; Shen, B.; Xiao, S.-Y.; Liu, X. Paneth Cells in Colonic Adenomas. Am. J. Surg. Pathol. 2013, 37, 98–103. [Google Scholar] [CrossRef]
- Atreya, I.; Neurath, M.F. Immune cells in colorectal cancer: Prognostic relevance and therapeutic strategies. Expert Rev. Anticancer Ther. 2008, 8, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Mariani, F.; Sena, P.; Roncucci, L. Inflammatory pathways in the early steps of colorectal cancer development. World J. Gastroenterol. 2014, 20, 9716–9731. [Google Scholar] [CrossRef]
- Burkholder, B.; Huang, R.-Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.-Q.; Gao, C.-Y.; Wang, B.-L.; Zhang, Y.-M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 182–201. [Google Scholar] [CrossRef] [Green Version]
- Sillo, T.O.; Beggs, A.D.; Morton, D.G.; Middleton, G. Mechanisms of immunogenicity in colorectal cancer. Br. J. Surg. 2019, 106, 1283–1297. [Google Scholar] [CrossRef]
- Väyrynen, J.P.; Tuomisto, A.; Klintrup, K.; Mäkelä, J.; Karttunen, T.J.; Mäkinen, M.J. Detailed analysis of inflammatory cell infiltration in colorectal cancer. Br. J. Cancer 2013, 109, 1839–1847. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, C.; Correale, C.; Gandelli, A.; Spinelli, A.; Dejana, E.; D’Alessio, S.; Danese, S. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 2015, 148, 1438–1451. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, C.; Ungaro, F.; Correale, C.; Arena, V.; Massimino, L.; Detmar, M.; Spinelli, A.; Carvello, M.; Mazzone, M.; Oliveira, A.I.; et al. Activation of the VEGFC/VEGFR3 Pathway Induces Tumor Immune Escape in Colorectal Cancer. Cancer Res. 2019, 79, 4196–4210. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungaro, F.; Colombo, P.; Massimino, L.; Ugolini, G.S.; Correale, C.; Rasponi, M.; Garlatti, V.; Rubbino, F.; Tacconi, C.; Spaggiari, P.; et al. Lymphatic endothelium contributes to colorectal cancer growth via the soluble matrisome component GDF11. Int. J. Cancer 2019, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Mathonnet, M. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J. Gastroenterol. 2014, 20, 4189. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4, 4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J. Mesenchymal Stem Cells in Cancer. Stem Cell Rev. Rep. 2008, 4, 119–124. [Google Scholar] [CrossRef]
- Liu, Y.; Han, Z.; Zhang, S.; Jing, Y.; Bu, X.; Wang, C.; Sun, K.; Jiang, G.; Zhao, X.; Li, R.; et al. Effects of Inflammatory Factors on Mesenchymal Stem Cells and Their Role in the Promotion of Tumor Angiogenesis in Colon Cancer. J. Biol. Chem. 2011, 286, 25007–25015. [Google Scholar] [CrossRef] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 2008, 118, 560–570. [Google Scholar]
- Kitakata, H.; Nemoto-Sasaki, Y.; Takahashi, Y.; Kondo, T.; Mai, M.; Mukaida, N. Essential roles of tumor necrosis factor receptor p55 in liver metastasis of intrasplenic administration of colon 26 cells. Cancer Res. 2002, 62, 6682–6687. [Google Scholar]
- De Simone, V.; Ronchetti, G.; Franzè, E.; Colantoni, A.; Ortenzi, A.; Fantini, M.C.; Rizzo, A.; Sica, G.S.; Sileri, P.; Rossi, P.; et al. Interleukin-21 sustains inflammatory signals that contribute to sporadic colon tumorigenesis. Oncotarget 2015, 6, 9908–9923. [Google Scholar] [CrossRef] [PubMed]
- Blatner, N.R.; Mulcahy, M.F.; Dennis, K.L.; Scholtens, D.; Bentrem, D.J.; Phillips, J.D.; Ham, S.; Sandall, B.P.; Khan, M.W.; Mahvi, D.M.; et al. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 2012, 4, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Zhou, R.; Liu, C.; Wang, Y.; Zhan, W.; Shao, Z.; Liu, J.; Zhang, F.; Xu, L.; Zhou, X.; et al. MicroRNA-105 is involved in TNF-α-related tumor microenvironment enhanced colorectal cancer progression. Cell Death Dis. 2017, 8, 3213. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.P.; Greer, P.K. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLoS ONE 2012, 7, 41797. [Google Scholar] [CrossRef]
- Qin, Z. Expression of tumor necrosis factor by different tumor cell lines results either in tumor suppression or augmented metastasis. J. Exp. Med. 1993, 178, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzinska-Ustymowicz, K.; Kemona, A. Transforming growth factor beta can be a parameter of aggressiveness of pT1 colorectal cancer. World J. Gastroenterol. 2005, 11, 1193–1195. [Google Scholar] [CrossRef]
- Bellam, N.; Pasche, B. TGF-β Signaling Alterations and Colon Cancer. Cancer Treat Res. 2010, 155, 85–103. [Google Scholar]
- Villalba, M.; Evans, S.R.; Vidal-Vanaclocha, F.; Calvo, A. Role of TGF-β in metastatic colon cancer: It is finally time for targeted therapy. Cell Tissue Res. 2017, 370, 29–39. [Google Scholar] [CrossRef]
- Zubeldia, I.G.; Bleau, A.-M.; Redrado, M.; Serrano, D.; Agliano, A.; Gil-Puig, C.; Vidal-Vanaclocha, F.; Lecanda, J.; Calvo, A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1-targeting peptides P17 and P144. Exp. Cell Res. 2013, 319, 12–22. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.; Samowitz, W.S.; Wolff, R.K.; Stevens, J.R.; Herrick, J.S. The NF-κB signalling pathway in colorectal cancer: Associations between dysregulated gene and miRNA expression. J. Cancer Res. Clin. Oncol. 2018, 144, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, K.; Han, G.-C.; Wang, R.-X.; Xiao, H.; Hou, C.-M.; Guo, R.-F.; Dou, Y.; Shen, B.-F.; Li, Y.; et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014, 7, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Elaraj, D.M. The Role of Interleukin 1 in Growth and Metastasis of Human Cancer Xenografts. Clin. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.Z.; Tsilidis, K.K.; Smith, M.W.; Hoffman-Bolton, J.; Visvanathan, K.; Platz, E.A.; Joshu, C.E. Polymorphisms in genes related to inflammation and obesity and colorectal adenoma risk. Mol. Carcinog. 2018, 57, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Komoda, H.; Tanaka, Y.; Honda, M.; Matsuo, Y.; Hazama, K.; Takao, T. Interleukin-6 Levels in Colorectal Cancer Tissues. World J. Surg. 1998, 22, 895–898. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S.; et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Yaoita, T.; Sasaki, Y.; Yokozawa, J.; Sato, T.; Kanno, N.; Sakuta, K.; Yagi, M.; Yoshizawa, K.; Iwano, D.; Nagino, K.; et al. Treatment with anti-interleukin-6 receptor antibody ameliorates intestinal polyposis in Apc(Min/+) mice under high-fat diet conditions. Tohoku J. Exp. Med. 2015, 235, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Lai, W.; Zhang, Y.; Liu, L.; Luo, X.; Zeng, Y.; Wu, H.; Lan, Q.; Chu, Z. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer 2014, 14, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Wang, W.; Tian, J.; Yin, K.; Tang, X.; Ma, J.; Xu, H.; Wang, S. IL-17A produced by peritoneal macrophages promote the accumulation and function of granulocytic myeloid-derived suppressor cells in the development of colitis-associated cancer. Tumor Biol. 2016, 37, 15883–15891. [Google Scholar] [CrossRef]
- Le Gouvello, S.; Bastuji-Garin, S.; Aloulou, N.; Mansour, H.; Chaumette, M.-T.; Berrehar, F.; Seikour, A.; Charachon, A.; Karoui, M.; Leroy, K.; et al. High prevalence of Foxp3 and IL17 in MMR-proficient colorectal carcinomas. Gut 2008, 57, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, K.; Kohama, H.; Umemura, M.; Uyttenhove, C.; Inagaki-Ohara, K.; Arakawa, T.; Harada, M.; Nakae, S.; Iwakura, Y.; Nishimaki, T.; et al. Interleukin-17A is involved in enhancement of tumor progression in murine intestine. Immunobiology 2012, 217, 54–60. [Google Scholar] [CrossRef]
- Hyun, Y.S.; Han, D.S.; Lee, A.R.; Eun, C.S.; Youn, J.; Kim, H.-Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef]
- Chae, W.-J.; Gibson, T.F.; Zelterman, D.; Hao, L.; Henegariu, O.; Bothwell, A.L.M. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 5540–5544. [Google Scholar] [CrossRef] [Green Version]
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Tuncer, S.; Banerjee, S. Eicosanoid pathway in colorectal cancer: Recent updates. World J. Gastroenterol. 2015, 21, 11748–11766. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Elsbach, P.; Weiss, J. Role of the bactericidal/permeability-increasing protein in host defence. Curr. Opin. Immunol. 1998, 10, 45–49. [Google Scholar] [CrossRef]
- Campbell, E.L.; MacManus, C.F.; Kominsky, D.J.; Keely, S.; Glover, L.E.; Bowers, B.E.; Scully, M.; Bruyninckx, W.J.; Colgan, S.P. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc. Natl. Acad. Sci. USA 2010, 107, 14298–14303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, J.; Godson, C.; Brady, H.R.; Macmathuna, P. Lipoxins: Pro-resolution lipid mediators in intestinal inflammation. Gastroenterology 2003, 124, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting Edge: Lipoxins Rapidly Stimulate Nonphlogistic Phagocytosis of Apoptotic Neutrophils by Monocyte-Derived Macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef]
- Marcon, R.; Bento, A.F.; Dutra, R.C.; Bicca, M.A.; Leite, D.F.P.; Calixto, J.B. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J. Immunol. 2013, 191, 4288–4298. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, Inflammation and Colorectal Cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef] [Green Version]
- Abel, S.; Riedel, S.; Gelderblom, W.C.A. Dietary PUFA and cancer. Proc. Nutr. Soc. 2014, 73, 361–367. [Google Scholar] [CrossRef]
- Sasazuki, S.; Inoue, M.; Iwasaki, M.; Sawada, N.; Shimazu, T.; Yamaji, T.; Takachi, R.; Tsugane, S. Intake of n-3 and n-6 polyunsaturated fatty acids and development of colorectal cancer by subsite: Japan Public Health Center-based prospective study. Int. J. Cancer 2011, 129, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Norat, T.; Bingham, S.; Ferrari, P.; Slimani, N.; Jenab, M.; Mazuir, M.; Overvad, K.; Olsen, A.; Tjønneland, A.; Clavel, F.; et al. Meat, Fish, and Colorectal Cancer Risk: The European Prospective Investigation into Cancer and Nutrition. JNCI J. Natl. Cancer Inst. 2005, 97, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Chavarro, J.E.; Lee, I.-M.; Willett, W.C.; Ma, J. A 22-year Prospective Study of Fish, n-3 Fatty Acid Intake, and Colorectal Cancer Risk in Men. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1136–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aglago, E.K.; Huybrechts, I.; Murphy, N.; Casagrande, C.; Nicolas, G.; Pischon, T.; Fedirko, V.; Severi, G.; Boutron-Ruault, M.-C.; Fournier, A.; et al. Consumption of Fish and Long-chain n-3 Polyunsaturated Fatty Acids Is Associated With Reduced Risk of Colorectal Cancer in a Large European Cohort. Clin. Gastroenterol. Hepatol. 2020, 18, 654–666. [Google Scholar] [CrossRef]
- Tutino, V.; De Nunzio, V.; Caruso, M.G.; Veronese, N.; Lorusso, D.; Di Masi, M.; Benedetto, M.L.; Notarnicola, M. Elevated AA/EPA Ratio Represents an Inflammatory Biomarker in Tumor Tissue of Metastatic Colorectal Cancer Patients. Int. J. Mol. Sci. 2019, 20, 2050. [Google Scholar] [CrossRef] [Green Version]
- Hull, M.A.; Sprange, K.; Hepburn, T.; Tan, W.; Shafayat, A.; Rees, C.J.; Clifford, G.; Logan, R.F.; Loadman, P.M.; Williams, E.A.; et al. Eicosapentaenoic acid and aspirin, alone and in combination, for the prevention of colorectal adenomas (seAFOod Polyp Prevention trial): A multicentre, randomised, double-blind, placebo-controlled, 2 × 2 factorial trial. Lancet 2018, 392, 2583–2594. [Google Scholar] [CrossRef] [Green Version]
- Shin, A.; Cho, S.; Sandin, S.; Lof, M.; Oh, M.Y.; Weiderpass, E. Omega-3 and -6 Fatty Acid Intake and Colorectal Cancer Risk in Swedish Women’s Lifestyle and Health Cohort. Cancer Res. Treat. 2020, 52, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Kantor, E.D.; Lampe, J.W.; Peters, U.; Vaughan, T.L.; White, E. Long-Chain Omega-3 Polyunsaturated Fatty Acid Intake and Risk of Colorectal Cancer. Nutr. Cancer 2014, 66, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Mocellin, M.C.; Silva, J.D.A.P.E.; Camargo, C.D.Q.; Fabre, M.E.D.S.; Gevaerd, S.; Naliwaiko, K.; Moreno, Y.M.F.; Nunes, E.A.; Trindade, E.B.S.D.M. Fish Oil Decreases C-Reactive Protein/Albumin Ratio Improving Nutritional Prognosis and Plasma Fatty Acid Profile in Colorectal Cancer Patients. Lipids 2013, 48, 879–888. [Google Scholar] [CrossRef]
- Silva, J.D.A.P.; Trindade, E.B.S.D.M.; Fabre, M.E.D.S.; Menegotto, V.M.; Gevaerd, S.; Buss, Z.D.S.; Frode, T.S. Fish Oil Supplement Alters Markers of Inflammatory and Nutritional Status in Colorectal Cancer Patients. Nutr. Cancer 2012, 64, 267–273. [Google Scholar] [CrossRef]
- Khankari, N.K.; Banbury, B.L.; Borges, M.C.; Haycock, P.; Albanes, D.; Arndt, V.; Berndt, S.I.; Bézieau, S.; Brenner, H.; Campbell, P.T.; et al. Mendelian Randomization of Circulating Polyunsaturated Fatty Acids and Colorectal Cancer Risk. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbalho, S.M.; de Alvares Goulart, R.; Quesada, K.; Bechara, M.D.; de Carvalho, A.d.C.A. Inflammatory bowel disease: Can omega-3 fatty acids really help. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2016, 29, 37–43. [Google Scholar]
- Mocellin, M.C.; Camargo, C.Q.; Nunes, E.A.; Fiates, G.M.R.; Trindade, E.B.S.M. A systematic review and meta-analysis of the n-3 polyunsaturated fatty acids effects on inflammatory markers in colorectal cancer. Clin. Nutr. 2016, 35, 359–369. [Google Scholar] [CrossRef]
- Gupta, R.A.; DuBois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Hull, M.A.; Ko, S.C.W.; Hawcroft, G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol. Cancer Ther. 2004, 3, 1031–1039. [Google Scholar] [PubMed]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of Intestinal Polyposis in ApcΔ716 Knockout Mice by Inhibition of Cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Calviello, G.; Di Nicuolo, F.; Gragnoli, S.; Piccioni, E.; Serini, S.; Maggiano, N.; Tringali, G.; Navarra, P.; Ranelletti, F.O.; Palozza, P. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE 2 induced ERK-1 and -2 and HIF-1α induction pathway. Carcinogenesis 2004, 25, 2303–2310. [Google Scholar] [CrossRef] [Green Version]
- Habbel, P.; Weylandt, K.H.; Lichopoj, K.; Nowak, J.; Purschke, M.; Wang, J.-D.; He, C.-W.; Baumgart, D.C.; Kang, J.X. Docosahexaenoic acid suppresses arachidonic acid-induced proliferation of LS-174T human colon carcinoma cells. World J. Gastroenterol. 2009, 15, 1079. [Google Scholar] [CrossRef]
- Fluckiger, A.; Dumont, A.; Derangère, V.; Rébé, C.; de Rosny, C.; Causse, S.; Thomas, C.; Apetoh, L.; Hichami, A.; Ghiringhelli, F.; et al. Inhibition of colon cancer growth by docosahexaenoic acid involves autocrine production of TNFα. Oncogene 2016, 35, 4611–4622. [Google Scholar] [CrossRef]
- Siddiqui, R.A.; Harvey, K.; Stillwell, W. Anticancer properties of oxidation products of docosahexaenoic acid. Chem. Phys. Lipids 2008, 153, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Sarabi, M.M.; Khorramabadi, R.M.; Zare, Z.; Eftekhar, E. Polyunsaturated fatty acids and DNA methylation in colorectal cancer. World J. Clin. Cases 2019, 7, 4172–4185. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, L.; Zhang, B.; He, M.; Dong, X.; Lin, X.; Jia, C.; Bai, X.; Dai, Y.; Su, Y.; et al. Elevation of n-3/n-6 PUFAs ratio suppresses mTORC1 and prevents colorectal carcinogenesis associated with APC mutation. Oncotarget 2016, 7, 76944–76954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, M.; Notarnicola, M.; Caruso, M.G.; Scavo, M.P.; Viggiani, M.T.; Tutino, V.; Polimeno, L.; Pesetti, B.; Di Leo, A.; Francavilla, A. Olive oil and omega-3 polyunsaturated fatty acids suppress intestinal polyp growth by modulating the apoptotic process in ApcMin/+ mice. Carcinogenesis 2014, 35, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-M.; Jeong, M.; Park, J.-M.; Kim, M.-Y.; Go, E.-J.; Cha, J.Y.; Kim, K.J.; Hahm, K.B. The ω-3 polyunsaturated fatty acids prevented colitis-associated carcinogenesis through blocking dissociation of β-catenin complex, inhibiting COX-2 through repressing NF-κB, and inducing 15-prostaglandin dehydrogenase. Oncotarget 2016, 7, 63583–63595. [Google Scholar] [PubMed] [Green Version]
- Piazzi, G.; D’Argenio, G.; Prossomariti, A.; Lembo, V.; Mazzone, G.; Candela, M.; Biagi, E.; Brigidi, P.; Vitaglione, P.; Fogliano, V.; et al. Eicosapentaenoic acid free fatty acid prevents and suppresses colonic neoplasia in colitis-associated colorectal cancer acting on Notch signaling and gut microbiota. Int. J. Cancer 2014, 135, 2004–2013. [Google Scholar] [CrossRef]
- Hawcroft, G.; Volpato, M.; Marston, G.; Ingram, N.; Perry, S.; Cockbain, A.; Race, A.; Munarini, A.; Belluzzi, A.; Loadman, P.; et al. The omega-3 polyunsaturated fatty acid eicosapentaenoic acid inhibits mouse MC-26 colorectal cancer cell liver metastasis via inhibition of PGE2-dependent cell motility. Br. J. Pharmacol. 2012, 166, 1724–1737. [Google Scholar] [CrossRef] [Green Version]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef]
- Fedirko, V.; McKeown-Eyssen, G.; Serhan, C.N.; Barry, E.L.; Sandler, R.S.; Figueiredo, J.C.; Ahnen, D.J.; Bresalier, R.S.; Robertson, D.J.; Anderson, C.W.; et al. Plasma lipoxin A 4 and resolvin D1 are not associated with reduced adenoma risk in a randomized trial of aspirin to prevent colon adenomas. Mol. Carcinog. 2017, 56, 1977–1983. [Google Scholar] [CrossRef]
- Zhuang, Q.; Meng, Q.; Xi, Q.; Wu, G. Association of serum inflammatory cytokines and Resolvin D1 concentration with pathological stage of colon cancer. Chin. J. Gastrointest. Surg. 2018, 21, 1285–1290. [Google Scholar]
- Zhong, X.; Lee, H.-N.; Surh, Y.-J. RvD1 inhibits TNFα-induced c-Myc expression in normal intestinal epithelial cells and destabilizes hyper-expressed c-Myc in colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Irún, P.; Lanas, A.; Piazuelo, E. Omega-3 Polyunsaturated Fatty Acids and Their Bioactive Metabolites in Gastrointestinal Malignancies Related to Unresolved Inflammation. A Review. Front. Pharmacol. 2019, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Gu, Y.; Tao, X.; Serhan, C.N.; Ji, R.-R. Resolvin D5 Inhibits Neuropathic and Inflammatory Pain in Male But Not Female Mice: Distinct Actions of D-Series Resolvins in Chemotherapy-Induced Peripheral Neuropathy. Front. Pharmacol. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, J.; Nimiya, Y.; Lee, K.S.S.; Sanidad, K.; Qi, W.; Sukamtoh, E.; Park, Y.; Liu, Z.; Zhang, G. ω-3 Polyunsaturated fatty acids and their cytochrome P450-derived metabolites suppress colorectal tumor development in mice. J. Nutr. Biochem. 2017, 48, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wan, M.; Huang, W.; Stanton, R.C.; Xu, Y. Maresins: Specialized Proresolving Lipid Mediators and Their Potential Role in Inflammatory-Related Diseases. Mediators Inflamm. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 397–413. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dalli, J.; Chiang, N.; Baron, R.M.; Quintana, C.; Serhan, C.N. Plasticity of Leukocytic Exudates in Resolving Acute Inflammation Is Regulated by MicroRNA and Proresolving Mediators. Immunity 2013, 39, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Wu, Y.; Zhao, F.; Wang, J. Maresin 1 Ameliorates Lung Ischemia/Reperfusion Injury by Suppressing Oxidative Stress via Activation of the Nrf-2-Mediated HO-1 Signaling Pathway. Oxid. Med. Cell. Longev. 2017, 2017, 9634803. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Ma, Z.; Ma, M.; Wang, D.; Xie, G.; Yin, Y.; Zhang, P.; Tao, K. Maresin 1 Mitigates Inflammatory Response and Protects Mice from Sepsis. Mediators Inflamm. 2016, 2016, 3798465. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, Q.; Tang, K.; Sun, Y.; Zhang, K.; Zhang, Y.; Luo, S.; Zhang, H.; Ye, D.; Huang, B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 2015, 364, 118–124. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, H.; Ni, X.; Shen, S.; Das, U.N. Growth Inhibitory Effect of Polyunsaturated Fatty Acids (PUFAs) on Colon Cancer Cells via Their Growth Inhibitory Metabolites and Fatty Acid Composition Changes. PLoS ONE 2015, 10, 0123256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zeng, J.; Huang, W.; Xu, Q.; Ye, D.; Sun, R.; Zhang, D. Colorectal Cancer Is Associated with a Deficiency of Lipoxin A 4, an Endogenous Anti-inflammatory Mediator. J. Cancer 2019, 10, 4719–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Kashihara, D.; Ichimura, A.; Kimura, I.; Tsujimoto, G.; Hirasawa, A. Role of free fatty acid receptors in the regulation of energy metabolism. Biochim. Biophys. Acta–Mol. Cell Biol. Lipids 2014, 1841, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Meier, K.E. Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology. Handb. Exp. Pharmacol. 2017, 236, 233–251. [Google Scholar] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Senatorov, I.S.; Moniri, N.H. The role of free-fatty acid receptor-4 (FFA4) in human cancers and cancer cell lines. Biochem. Pharmacol. 2018, 150, 170–180. [Google Scholar] [CrossRef]
- Nakashima, C.; Shingo, K.; Fujiwara-Tani, R.; Luo, Y.; Kawahara, I.; Goto, K.; Sasaki, T.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Expression of long-chain fatty acid receptor GPR40 is associated with cancer progression in colorectal cancer: A retrospective study. Oncol. Lett. 2018, 15, 8641–8646. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wang, H.; Zhao, X.; Shi, Y.; Jin, M.; Wan, B.; Xu, H.; Cheng, Y.; Ge, H.; Zhang, Y. Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene 2013, 32, 5541–5550. [Google Scholar] [CrossRef]
- Liu, Z.; Hopkins, M.M.; Zhang, Z.; Quisenberry, C.B.; Fix, L.C.; Galvan, B.M.; Meier, K.E. Omega-3 Fatty Acids and Other FFA4 Agonists Inhibit Growth Factor Signaling in Human Prostate Cancer Cells. J. Pharmacol. Exp. Ther. 2015, 352, 380–394. [Google Scholar] [CrossRef]
- Hopkins, M.; Zhang, Z.; Liu, Z.; Meier, K. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J. Clin. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Hu, Z.; Qi, H.; Shi, Z.; Chang, Y.; Yao, Q.; Cui, H.; Zheng, L.; Han, Y.; Han, X.; et al. G-protein-coupled receptors mediate ω-3 PUFAs-inhibited colorectal cancer by activating the Hippo pathway. Oncotarget 2016, 7, 58315–58330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobraten, K.; Haug, T.M.; Kleiveland, C.R.; Lea, T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis. 2013, 12, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clària, J.; Lee, M.H.; Serhan, C.N. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med. 1996, 2, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, Foods, and Colorectal Cancer Prevention. Gastroenterology 2015, 148, 1244–1260. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhang, L.; Xu, H.-J.; Li, Y.; Hu, C.-M.; Yang, J.-Y.; Sun, M.-Y. The Anti-Inflammatory Effects of Vitamin D in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2736. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, H.; Murakami, S.; Suzuki, S.; Kudo, H.; Sassa, S.; Sakamoto, S. Chemopreventive effect of a vitamin D3 analog, alfacalcidol, on colorectal carcinogenesis in mice with ulcerative colitis. Anticancer. Drugs 2007, 18, 1183–1187. [Google Scholar] [CrossRef]
- Hummel, D.M.; Thiem, U.; Höbaus, J.; Mesteri, I.; Gober, L.; Stremnitzer, C.; Graça, J.; Obermayer-Pietsch, B.; Kallay, E. Prevention of preneoplastic lesions by dietary vitamin D in a mouse model of colorectal carcinogenesis. J. Steroid Biochem. Mol. Biol. 2013, 136, 284–288. [Google Scholar]
- Ungprasert, P.; Cheungpasitporn, W.; Crowson, C.S.; Matteson, E.L. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur. J. Intern. Med. 2015, 26, 285–291. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Study Reference | Type of Study | Outcome |
|---|---|---|
| Sasazuki et al, 2011; Norat et al, 2005; Hall et al, 2008; Aglago et al., 2019 [55,56,57,58] | Clinical prospective studies | ω-3/CRC risk inverse correlation |
| Tutino et al., 2019 [58] | Clinical observational study | High ratio of ω-6/ω-3 as metastatic CRC biomarker |
| Hull et al., 2018 [59] | Clinical, multicentre, randomized, double-blind, placebo-controlled, 2 × 2 factorial trial | EPA does not reduce CRC risk |
| Shin et al., 2020 [60] | Clinical prospective study | DHA intake reduces CRC risk |
| Khankari et al., 2020 [61] | Clinical observational study | Shorter-chain PUFAs→reduced CRC risk; longer-chain PUFA levels→increased CRC risk |
| Oshima et al., 1996 [64] | Preclinical study on APCΔ716 mouse model | COX-2 derived AA lipids are essential for tumor growth |
| Calviello et al., 2004 [65] | In vitro study on colon cancer cells | EPA and DHA block COX-2 expression |
| Fluckiger et al., 2016; Siddiqui et al., 2008 [67,68] | In vitro study on colon cancer cells | DHA blocks AA-induced proliferation and apoptosis |
| Sarabi et al., 2019 [69] | Preclinical study on APCmin/+ mouse model and in vitro study on colon cancer cells | ω-3 PUFAs enhance apoptosis via specific promoter methylation |
| Liu et al., 2016 [70] | Preclinical study on APCmin/+ mouse model | ω-3 PUFAs repressed tumor growth and burden |
| Barone et al., 2014 [71] | Preclinical study on APCmin/+ mouse model | ω-3 PUFAs repressed tumor growth via apoptosis inhibition |
| Han et al., 2016 [72] | In vitro study on colon cancer cells and preclinical study on AOM/DSS-treated mice | DHA induces apoptosis and reduces tumor growth |
| Piazzi et al., 2014 [73] | Preclinical study on AOM/DSS-treated mice | EPA reduces tumor growth by restoring Notch signaling |
| Hawcroft et al., 2012 [74] | Preclinical study by intrasplenic injection of mouse colon cancer cells | EPA reduces liver metastasis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungaro, F.; D’Alessio, S.; Danese, S. The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies. Cancers 2020, 12, 2060. https://doi.org/10.3390/cancers12082060
Ungaro F, D’Alessio S, Danese S. The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies. Cancers. 2020; 12(8):2060. https://doi.org/10.3390/cancers12082060
Chicago/Turabian StyleUngaro, Federica, Silvia D’Alessio, and Silvio Danese. 2020. "The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies" Cancers 12, no. 8: 2060. https://doi.org/10.3390/cancers12082060
APA StyleUngaro, F., D’Alessio, S., & Danese, S. (2020). The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies. Cancers, 12(8), 2060. https://doi.org/10.3390/cancers12082060