DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The DNA Damage Response
2.1. Types of DNA Damage
2.2. DNA Damage Repair Pathways
3. DNA Damage-Inducing Therapies
3.1. Radiotherapy
3.1.1. External Beam Radiotherapy
3.1.2. Brachytherapy
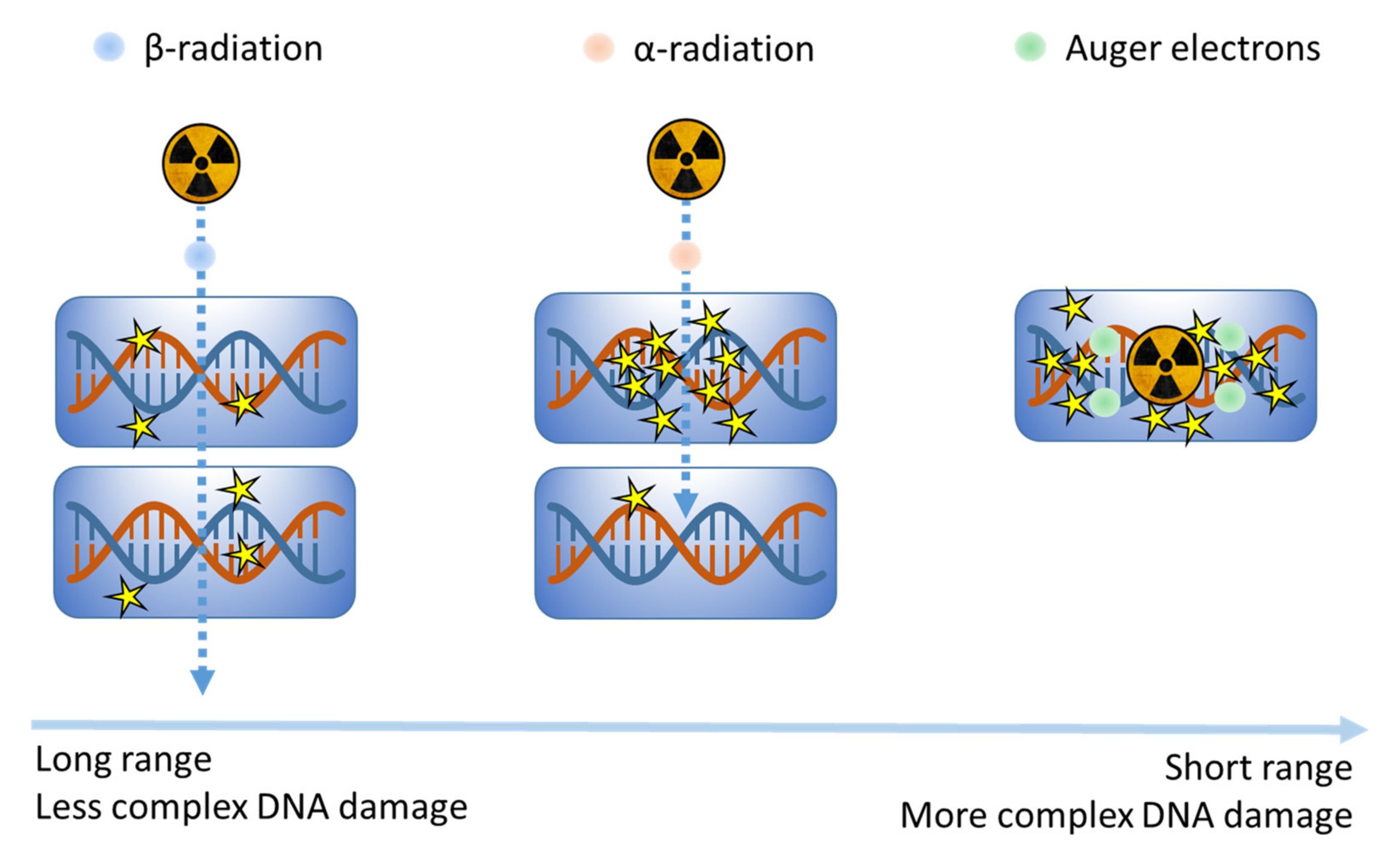
3.1.3. Molecular Radionuclide Therapy (MRT)
3.2. Cytotoxic Chemotherapy
3.2.1. Alkylating Agents and Platinum-Based Compounds
3.2.2. Antimetabolites
3.2.3. Topoisomerase Inhibitors
3.2.4. Antitumor Antibiotics
3.2.5. Improvement of Chemotherapy
3.3. Targeted Therapies: Modulators of the DDR
4. Combination Approaches Involving DNA Damage-Inducing Therapies
4.1. Radiotherapy and Chemotherapy Combinations
4.2. DDR Modulator Combinations
4.3. Immunotherapy Combinations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pilié, P.; Tang, C.; Mills, G.; Yap, T. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Yixin Yao, W.D. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1–3. [Google Scholar]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Gianfaldoni, S.; Gianfaldoni, R.; Wollina, U.; Lotti, J.; Tchernev, G.; Lotti, T. An overview on radiotherapy: From its history to its current applications in dermatology. Open Access Maced. J. Med. Sci. 2017, 5, 521–525. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Prim. 2019, 5, 13. [Google Scholar] [CrossRef]
- Redmond, K.M.; Wilson, T.R.; Johnston, P.G.; Longley, D.B. Resistance mechanisms to cancer chemotherapy. Front. Biosci. 2008, 13, 5138–5154. [Google Scholar] [CrossRef]
- Dexheimer, T. DNA repair Pathways and Mechanisms. In DNA Repair of Cancer Stem Cells; Springer: Berlin/Heidelberg, Germany, 2014; pp. 19–32. ISBN 9789400745902. [Google Scholar]
- Cannan, W.; Pederson, D. Mechanisms and Consequences of Double-strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Frankenberg-Schwager, M.; Frankenberg, D. DNA double-Strand breaks: Their repair and relationship to cell killing in yeast. Int. J. Radiat. Biol. 1990, 58, 569–575. [Google Scholar] [CrossRef]
- Pfeiffer, P. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [PubMed]
- Clingen, P.H.; Arlett, C.F.; Roza, L.; Mori, T.; Nikaido, O.; Green, M.H.L. Induction of Cyclobutane Pyrimidine Dimers, Pyrimidine(6-4)pyrimidone Photoproducts, and Dewar Valence Isomers by Natural Sunlight in Normal Human Mononuclear Cells. Cancer Res. 1995, 55, 2245–2248. [Google Scholar]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb Perspect Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Sengerová, B.; Wang, A.T.; Mchugh, P.J. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle 2011, 10, 3999–4008. [Google Scholar] [CrossRef]
- Budzowska, M.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511. [Google Scholar]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.L.; Zubizarreta, E.; Bray, F.; Ferlay, J.; Barton, M. Global Access to Radiotherapy Services: Have We Made Progress During the Past Decade? J. Glob. Oncol. 2016, 2, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E. Recent developments in radiotherapy. N. Engl. J. Med. 2017, 377, 1065–1075. [Google Scholar] [CrossRef]
- Vandyk, J.; Battista, J. Cobalt-60: An old modality, a renewed challenge. Curr. Oncol. 1995, 3, 8–17. [Google Scholar]
- Tian, X.; Liu, K.; Hou, Y.; Cheng, J.; Zhang, J. The evolution of proton beam therapy: Current and future status (Review). Mol. Clin. Oncol. 2017, 8, 15–21. [Google Scholar] [CrossRef]
- Kanderup, K.; Menard, C.; Polgar, C.; Lindegaard, J.; Kirisits, C.; Potter, R. Advancements in brachytherapy. Adv. Drug Deliv. Rev. 2017, 109, 15–25. [Google Scholar] [CrossRef]
- Nitipir, C.; Niculae, D.; Orlov, C.; Barbu, M.A.; Popescu, B.; Popa, A.M.; Pantea, A.M.S.; Stanciu, A.E.; Galateanu, B.; Ginghina, O.; et al. Update on radionuclide therapy in oncology. Oncol. Lett. 2017, 14, 7011–7015. [Google Scholar]
- Mohan, R.; Peeler, C.; Guan, F.; Bronk, L.; Cao, W.; Grosshans, D. Radiobiological issues in proton therapy. Acta Oncol. 2017, 56, 1363–1373. [Google Scholar] [CrossRef]
- Zaider, M.; Bardash, M.; Fung, A. Molecular damage induced directly and indirectly by ionizing radiation in DNA. Int. J. Radiat. Biol. 1994, 66, 459–465. [Google Scholar] [CrossRef]
- Gulston, M. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res. 2002, 30, 3464–3472. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Mladenov, E.; Mangelis, A.; Laskaratou, D.A.; Fragkoulis, G.I.; Hellweg, C.E.; Martin, O.A.; Emfietzoglou, D.; et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radic. Res. 2016, 50, S64–S78. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, T.A.; Palmer, P.; Maniou, Z.; Lomax, M.E.; O’Neill, P. Interplay of two major repair pathways in the processing of complex double-strand DNA breaks. DNA Repair (Amst.) 2008, 7, 1372–1383. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Sinclair, W.K.; Morton, R.A. X-Ray Sensitivity during the Cell Generation Cycle of Cultured Chinese Hamster Cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Vijayappa, S.; Kurimasa, A.; Ogawa, K.; Chen, D.J. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006, 66, 8352–8355. [Google Scholar] [CrossRef]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef]
- Forker, L.J.; Choudhury, A.; Kiltie, A.E. Biomarkers of Tumour Radiosensitivity and Predicting Benefit from Radiotherapy. Clin. Oncol. 2015, 27, 561–569. [Google Scholar] [CrossRef]
- Withers, H.R. Biologic basis for altered fractionation schemes. Cancer 1985, 55, 2086–2095. [Google Scholar] [CrossRef]
- Wilson, R. Radiological use of fast protons. Radiology 1946, 47, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Vitti, E.T.; Parsons, J.L. The radiobiological effects of proton beam therapy: Impact on DNA damage and repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Britten, R.A.; Nazaryan, V.; Davis, L.K.; Klein, S.B.; Nichiporov, D.; Mendonca, M.S.; Wolanski, M.; Nie, X.; George, J.; Keppel, C. Variations in the RBE for Cell Killing Along the Depth-Dose Profile of a Modulated Proton Therapy Beam. Radiat. Res. 2013, 179, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, J. Low-dose-rate or high-dose-rate brachytherapy in treatment of prostate cancer—Between options. J. Contemp. Brachyther. 2013, 5, 33–41. [Google Scholar] [CrossRef]
- Sun Myint, A.; Stewart, A.; Mills, J.; Sripadam, R.; Whitmarsh, K.; Roy, R.; Franklin, A.; Dhadda, A. Treatment: The role of contact X-ray brachytherapy (Papillon) in the management of early rectal cancer. Color. Dis. 2019, 21, 45–52. [Google Scholar] [CrossRef]
- Kaufman, S.A.; DiPetrillo, T.A.; Price, L.L.; Midle, J.B.; Wazer, D.E. Long-term outcome and toxicity in a Phase I/II trial using high-dose-rate multicatheter interstitial brachytherapy for T1/T2 breast cancer. Brachytherapy 2007, 6, 286–292. [Google Scholar] [CrossRef]
- Martinez, A.; Demanes, J.; Vargas, C.; Schour, L.; Ghilezan, M.; Gustafson, G. High-Dose-Rate Prostate Brachytherapy: An Excellent Accelerated-Hypofractionated Treatment for Favorable Prostate Cancer. Am. J. Clin. Oncol. 2010, 33, 481–488. [Google Scholar] [CrossRef]
- Lettmaier, S.; Kreppner, S.; Lotter, M.; Walser, M.; Ott, O.; Fietkau, R.; Strnad, V. Radiation exposure of the heart, lung and skin by radiation therapy for breast cancer: A dosimetric comparison between partial breast irradiation using multicatheter brachytherapy and whole breast teletherapy. Radiother. Oncol. 2011, 100, 189–194. [Google Scholar] [CrossRef]
- Chen, H.; Bao, Y.; Yu, L.; Jia, R.; Cheng, W.; Shao, C. Comparison of Cellular Response to low-dose-rate 125I seed irradiation and high-dose-rate gamma irradiation in human lung cancer cells. Brachytherapy 2012, 11, 149–156. [Google Scholar] [CrossRef]
- Carignan, D.; Lessard, T.; Villeneuve, L.; Desjardins, S.; Magnan, S.; Després, P.; Martin, A.G.; Foster, W.; Guillemette, C.; Lévesque, É.; et al. DNA repair gene polymorphisms, tumor control, and treatment toxicity in prostate cancer patients treated with permanent implant prostate brachytherapy. Prostate 2020, 80, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Memon, K.; Lewandowski, R.; Kulik, L.; Riaz, A.; Mulcahy, M.; Salem, R. Radioembolization for primary and metastatic liver cancer. Semin. Radiat. Oncol. 2011, 21, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Gates, V.L.; Atassi, B.; Lewandowski, R.J.; Mulcahy, M.F.; Ryu, R.K.; Sato, K.T.; Baker, T.; Kulik, L.; Gupta, R.; et al. Radiation segmentectomy: A novel approach to increase safety and efficacy of radioembolization. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sfakianakis, G.N.; DeLand, F.H. Radioimmunodiagnosis and Radioimmunotherapy. J. Nucl. Med. 1982, 23, 840–850. [Google Scholar]
- Bodei, L.; Kidd, M.; Paganelli, G.; Grana, C.M.; Drozdov, I.; Cremonesi, M.; Lepensky, C.; Kwekkeboom, D.J.; Baum, R.P.; Krenning, E.P.; et al. Long-term tolerability of PRRT in 807 patients with neuroendocrine tumours: The value and limitations of clinical factors. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 5–19. [Google Scholar] [CrossRef]
- Kassis, A. Therapeutic Radionuclides: Biophysical and Radiobiologic Principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef]
- Pouget, J.P.; Lozza, C.; Deshayes, E.; Boudousq, V.; Navarro-Teulon, I. Introduction to radiobiology of targeted radionuclide therapy. Front. Med. 2015, 2, 12. [Google Scholar] [CrossRef]
- Song, H.; Senthamizhchelvan, S.; Hobbs, R.F.; Sgouros, G. Alpha Particle Emitter Radiolabeled Antibody for Metastatic Cancer: What Can We Learn from Heavy Ion Beam Radiobiology? Antibodies 2012, 1, 124–148. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Hovingh, S.; Ten Cate, R.; Krawczyk, P.; Stap, J.; Hoebe, R.; Aten, J.; Barendsen, G.W. Relative biological effectiveness of high linear energy transfer α-particles for the induction of DNA-double-strand breaks, chromosome aberrations and reproductive cell death in SW-1573 lung tumour cells. Oncol. Rep. 2012, 27, 769–774. [Google Scholar] [CrossRef]
- Pouget, J.P.; Navarro-Teulon, I.; Bardiès, M.; Chouin, N.; Cartron, G.; Pèlegrin, A.; Azria, D. Clinical radioimmunotherapy-the role of radiobiology. Nat. Rev. Clin. Oncol. 2011, 8, 720–734. [Google Scholar] [CrossRef]
- Fondell, A.; Edwards, K.; Ickenstein, L.M.; Sjöberg, S.; Carlsson, J.; Gedda, L. Nuclisome: A novel concept for radionuclide therapy using targeting liposomes. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 114–123. [Google Scholar] [CrossRef]
- Hoefnagel, A. Radionuclide therapy of the thyroid. Nucl. Med. 1991, 18, 408–431. [Google Scholar]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 trial of 177lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, E.A.M.; Van Weerden, W.M.; Nonnekens, J.; De Jong, M. The future of PSMA-targeted radionuclide therapy: An overview of recent preclinical research. Pharmaceutics 2019, 11, 560. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Kersemans, V.; Allen, P.D.; Terry, S.Y.A.; Baguña Torres, J.; Mosley, M.; Smart, S.C.; Lee, B.Q.; Falzone, N.; Vallis, K.A.; et al. Imaging DNA Damage Repair in vivo Following 177 Lu-DOTATATE Therapy. J. Nucl. Med. 2019, 61, 743–750. [Google Scholar] [CrossRef]
- Feijtel, D.; de Jong, M.; Nonnekens, J. Peptide receptor radionuclide therapy: Looking back, looking forward. Curr. Top. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Goodman, L.; Wintrobe, M.; Dameshek, W.; Goodman, M.; Gilman, A.; McLennan, M. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 21, 126–132. [Google Scholar] [CrossRef]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef]
- Krishnan, B.; Morgan, G.J. Non-Hodgkin lymphoma secondary to cancer chemotherapy. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 377–380. [Google Scholar] [CrossRef]
- Wang, Y.; Probin, V.; Zhou, D. Cancer Therapy-Induced Residual Bone Marrow Injury: Mechanisms of Induction and Implication for Therapy. Curr. Cancer Ther. Rev. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of chemotherapeutic agents. Incidence, treatment and prevention. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef]
- Hansen, L.T.; Lundin, C.; Spang-Thomsen, M.; Petersen, L.N.; Helleday, T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int. J. Cancer 2003, 105, 472–479. [Google Scholar] [CrossRef]
- Drabløs, F.; Feyzi, E.; Aas, P.; Vaagbø, C.; Kavli, B.; Bratlie, M.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair (Amst.) 2004, 3, 1389–1407. [Google Scholar]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Samson, L.; Thomale, J.; Rajewsky, M.F. Alternative pathways for the in vivo repair of O6-alkylguanine and O4-alkylthymine in Escherichia coli: The adaptive response and nucleotide excision repair. EMBO J. 1988, 7, 2261–2267. [Google Scholar] [CrossRef]
- Wang, K.; Lu, J.; Li, R. The events that occur when cisplatin encounters cells. Coord. Chem. Rev. 1996, 151, 53–88. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum resistance in ovarian cancer: Role of DNA repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Fujinaka, Y.; Matsuoka, K.; Iimori, M.; Tuul, M.; Sakasai, R.; Yoshinaga, K.; Saeki, H.; Morita, M.; Kakeji, Y.; Gillespie, D.A.; et al. ATR-Chk1 signaling pathway and homologous recombinational repair protect cells from 5-fluorouracil cytotoxicity. DNA Repair (Amst.) 2012, 11, 247–258. [Google Scholar] [CrossRef]
- Kinsella, T.J. Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. 2009, 15, 1853–1859. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Wilson, D.M. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci. 2009, 66, 788–799. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA TOPOISOMERASES: Structure, Function and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Khadka, D.B.; Cho, W. Topoisomerase inhibitors as anticancer agents: A patent update. Expert Opin. Ther. Pat. 2013, 23, 1033–1056. [Google Scholar] [CrossRef]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar] [CrossRef]
- Avemann, K.; Knippers, R.; Koller, T.; Sogo, J.M. Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol. Cell. Biol. 1988, 8, 3026–3034. [Google Scholar] [CrossRef]
- John, L. Nitiss Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar]
- Xu, Y.; Her, C. Inhibition of topoisomerase (DNA) I (TOP1): DNA damage repair and anticancer therapy. Biomolecules 2015, 5, 1652–1670. [Google Scholar] [CrossRef]
- Nitiss, J.; Wang, J.C. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc. Natl. Acad. Sci. USA 1988, 85, 7501–7505. [Google Scholar] [CrossRef]
- De Campos-Nebel, M.; Larripa, I.; González-Cid, M. Topoisomerase ii-mediated DNA damage is differently repaired during the cell cycle by non-homologous end joining and homologous recombination. PLoS ONE 2010, 5, e12541. [Google Scholar] [CrossRef]
- Maede, Y.; Shimizu, H.; Fukushima, T.; Kogame, T.; Nakamura, T.; Miki, T.; Takeda, S.; Pommier, Y.; Murai, J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2013, 13, 214–220. [Google Scholar] [CrossRef]
- Cutts, S.M.; Nudelman, A.; Rephaeli, A.; Phillips, D.R. The power and potential of doxorubicin-DNA adducts. IUBMB Life 2005, 57, 73–81. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Sastry, M.; Fiala, R.; Lipman, R.; Tomasz, M.; Patel, D. Solution Structure of the Monoalkylated Mitomycin C-DNA Complex. J. Mol. Biol. 1995, 247, 338–359. [Google Scholar] [CrossRef]
- Povirk, L. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996, 355, 71–89. [Google Scholar] [CrossRef]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef]
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for improving tumor targetability and efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of adjuvant chemotherapy for stage III colon cancer. N. Engl. J. Med. 2018, 378, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Shen, L. Molecular targeted therapy of cancer: The progress and future prospect. Front. Lab. Med. 2017, 1, 69–75. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; David, S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Differential trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2013, 72, 5588–5599. [Google Scholar] [CrossRef]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020, 20, 55–73. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Reaper, P.; Griffiths, M.; Long, J.; Charrier, J.; MacCormick, S.; Charlton, P.; Golec, J.; Pollard, J. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.B.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Synthetic lethality screens point the way to new cancer drug targets. Nat. Rev. Drug Discov. 2017, 16, 589–591. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Naipal, K.A.T.; Verkaik, N.S.; Ameziane, N.; Van Deurzen, C.H.M.; Brugge, P.T.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef]
- Gunderson, C.C.; Moore, K.N. BRACAnalysis CDx as a companion diagnostic tool for Lynparza. Expert Rev. Mol. Diagn. 2015, 15, 1111–1116. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Glickman, M.S.; Sawyers, C.L. Converting cancer therapies into cures: Lessons from infectious diseases. Cell 2012, 148, 1089–1098. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Hennequin, C.; Favaudon, V. Biological basis for chemo-radiotherapy interactions. Eur. J. Cancer 2002, 38, 223–230. [Google Scholar] [CrossRef]
- Frei, E.; Holland, J.; Schneiderman, M.; Pinkel, D.; Selkirk, G.; Freireich, E.; Silver, R.; Gold, G.; Regelson, W. A comparative study of two regimens of combination chemotherapy in acute leukemia. Blood 1958, 13, 1126–1148. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Van Der Wilt, C.L.; Van Moorsel, C.J.A.; Kroep, J.R.; Bergman, A.M.; Ackland, S.P. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol. Ther. 2000, 87, 227–253. [Google Scholar] [CrossRef]
- Paccagnella, A.; Orlando, A.; Marchiori, C.; Zorat, P.L.; Cavaniglia, G.; Sileni, V.C.; Jirillo, A.; Tomio, L.; Fila, G.; Fede, A.; et al. Phase III trial of initial chemotherapy in stage III or IV head and neck cancers: A study by the gruppo di studio sui tumori della testa e del collo. J. Natl. Cancer Inst. 1994, 86, 265–272. [Google Scholar] [CrossRef]
- Vaccaro, V.; Sperduti, I.; Milella, M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 365, 768–769. [Google Scholar]
- Jaffe, N.; Paed, D.; Traggis, D.; Salian, S.; Cassady, J.R. Improved outlook for Ewing’s sarcoma with combination chemotherapy (vincristine, actinomycin D and cyclophosphamide) and radiation therapy. Cancer 1976, 38, 1925–1930. [Google Scholar] [CrossRef]
- Steel, G.G. Terminology in the description of drug-radiation interactions. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1145–1150. [Google Scholar] [CrossRef]
- Rose, P.; Bundy, B.; Watkins, E.; Thigpen, J.; Deppe, G.; Maiman, M.; Clarke-Pearson, D.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef]
- Boeckman, H.J.; Trego, K.S.; Turchi, J.J. Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol. Cancer Res. 2005, 3, 277–285. [Google Scholar] [CrossRef]
- Shewach, D.S.; Lawrence, T.S. Antimetabolite radiosensitizers. J. Clin. Oncol. 2007, 25, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Milosevic, M.; Fyles, A.; Pintilie, M.; Viswanathan, A.N. Trends in the utilization of brachytherapy in cervical cancer in the United States. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Aupérin, A.; Le Péchoux, C.; Rolland, E.; Curran, W.J.; Furuse, K.; Fournel, P.; Belderbos, J.; Clamon, G.; Ulutin, H.C.; Paulus, R.; et al. Meta-analysis of concomitant versus sequential radiochemotherapy in locally advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Chalmers, A.J. Science in Focus: Combining Radiotherapy with Inhibitors of the DNA Damage Response. Clin. Oncol. 2016, 28, 279–282. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Rajan, A.; Carter, C.A.; Kelly, R.J.; Gutierrez, M.; Kummar, S.; Szabo, E.; Yancey, M.A.; Ji, U.; Mannargudi, B.; Woo, S.; et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res. 2012, 18, 2344–2351. [Google Scholar] [CrossRef]
- Matulonis, U.; Monk, B. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: Does a development pathway forward exist? Ann. Oncol. 2017, 28, 443–447. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. B 2019, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol. 2014, 11, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, E.J.; De Ruysscher, D.K.; Pimentel, V.O.; Marcus, D.; Berbee, M.; Hoeben, A.; Rekers, N.; Theys, J.; Yaromina, A.; Dubois, L.J.; et al. Combining radiotherapy with immunotherapy: The past, the present and the future. Br. J. Radiol. 2017, 90, 6–8. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. https://doi.org/10.3390/cancers12082098
Reuvers TGA, Kanaar R, Nonnekens J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers. 2020; 12(8):2098. https://doi.org/10.3390/cancers12082098
Chicago/Turabian StyleReuvers, Thom G. A., Roland Kanaar, and Julie Nonnekens. 2020. "DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage" Cancers 12, no. 8: 2098. https://doi.org/10.3390/cancers12082098
APA StyleReuvers, T. G. A., Kanaar, R., & Nonnekens, J. (2020). DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers, 12(8), 2098. https://doi.org/10.3390/cancers12082098