Relation of Neutrophil Gelatinase-Associated Lipocalin Overexpression to the Resistance to Apoptosis of Tumor B Cells in Chronic Lymphocytic Leukemia

 , and
, and
Abstract
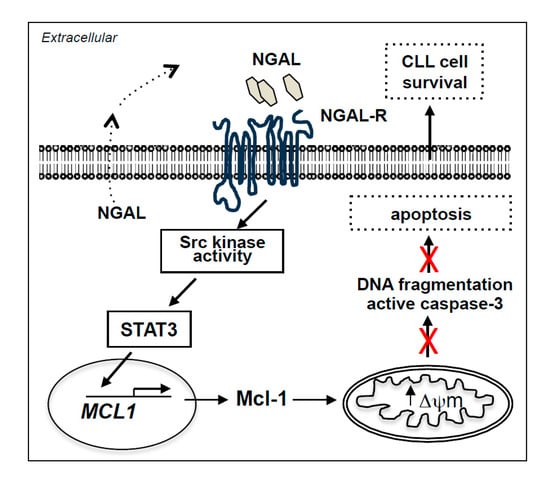
:
1. Introduction
2. Results
2.1. Serum NGAL Levels Are Elevated in Untreated Patients with CLL, and Return to Basal Levels When Patients Achieve Remission
2.2. The Expression of NGAL Reveals Differences between PBMCs from Normal Controls and Patients with CLL
2.3. NGAL-R Is Overexpressed in CLL Cells from Untreated Patients, and Downregulated in CLL Cells from Patients in Remission
2.4. Blocking NGAL-R Induces Death in NGAL-R+ CLL Cells
2.5. NGAL Protects CLL Cells from Spontaneous Death
2.6. NGAL Counteracts the Intrinsic Apoptosis Pathway in CLL Cells
2.7. NGAL Upregulates the Expression of Mcl-1 and STAT3 in CLL Cells
2.8. NGAL-Mediated CLL Cell Survival Involves the Src/STAT3/Mcl-1 Signaling Pathway
3. Discussion
4. Methods
4.1. Ethics Statement
4.2. Patients, Serum and Cell Separation
4.3. Cell Culture Conditions
4.4. ELISAs
4.5. Flow Cytometry
4.6. DNA Fragmentation Assay
4.7. Real Time PCR Assays
4.8. Immunoblotting
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pleyer, L.; Egle, A.; Hartmann, T.N.; Greil, R. Molecular and cellular mechanisms of CLL: Novel therapeutic approaches. Nat. Rev. Clin. Oncol. 2009, 6, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Burger, J.A. Cell Trafficking in Chronic Lymphocytic Leukemia. Open J. Hematol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Maffei, R.; Fiorcari, S.; Martinelli, S.; Potenza, L.; Luppi, M.; Marasca, R. Targeting neoplastic B cells and harnessing microenvironment: The “double face” of ibrutinib and idelalisib. J. Hematol. Oncol. 2015, 8, 60–82. [Google Scholar] [CrossRef] [Green Version]
- Pula, B.; Golos, A.; Gorniak, P.; Jamroziak, K. Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 1834. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Almasan, A. Development of venetoclax for therapy of lymphoid malignancies. Drug Des. Dev. Ther. 2017, 11, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.; Hallek, M. Venetoclax after idelalisib: Relevant progress for CLL. Blood 2018, 131, 1632–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta 2012, 1826, 129–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [PubMed]
- Bauvois, B.; Susin, S.A. Revisiting Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Cancer: Saint or Sinner? Cancers 2018, 10, 336. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Meschi, T.; Nouvenne, A.; Mattiuzzi, C.; Borghi, L. Neutrophil gelatinase-associated lipocalin in cancer. Adv. Clin. Chem. 2014, 64, 179–219. [Google Scholar] [PubMed]
- Virzi, G.M.; Clementi, A.; de Cal, M.; Cruz, D.N.; Ronco, C. Genomics and biological activity of neutrophil gelatinase-associated lipocalin in several clinical settings. Blood Purif. 2013, 35, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Makris, K.; Rizos, D.; Kafkas, N.; Haliassos, A. Neutrophil gelatinase-associated lipocalin as a new biomarker in laboratory medicine. Clin. Chem. Lab. Med. 2012, 50, 1519–1532. [Google Scholar] [CrossRef]
- Iqbal, N.; Choudhary, R.; Chan, J.; Wentworth, B.; Higginbotham, E.; Maisel, A.S. Neutrophil gelatinase-associated lipocalin as diagnostic and prognostic tool for cardiovascular disease and heart failure. Expert Opin. Med Diagn. 2013, 7, 209–220. [Google Scholar] [CrossRef]
- Paragas, N.; Qiu, A.; Hollmen, M.; Nickolas, T.L.; Devarajan, P.; Barasch, J. NGAL-Siderocalin in kidney disease. Biochim. Biophys. Acta 2012, 1823, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin: New paths for an old shuttle. Cancer Ther. 2007, 5, 463–470. [Google Scholar]
- Yang, J.; Moses, M.A. Lipocalin 2: A multifaceted modulator of human cancer. Cell Cycle 2009, 8, 2347–2352. [Google Scholar] [CrossRef]
- Bolignano, D.; Donato, V.; Lacquaniti, A.; Fazio, M.R.; Bono, C.; Coppolino, G.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: A new protein enters the scene. Cancer Lett. 2010, 288, 10–16. [Google Scholar] [CrossRef]
- Kamiguti, A.S.; Lee, E.S.; Till, K.J.; Harris, R.J.; Glenn, M.A.; Lin, K.; Chen, H.J.; Zuzel, M.; Cawley, J.C. The role of matrix metalloproteinase 9 in the pathogenesis of chronic lymphocytic leukaemia. Br. J. Haematol. 2004, 125, 128–140. [Google Scholar] [CrossRef]
- Bouchet, S.; Bauvois, B. Neutrophil Gelatinase-Associated Lipocalin (NGAL), Pro-Matrix Metalloproteinase-9 (pro-MMP-9) and Their Complex Pro-MMP-9/NGAL in Leukaemias. Cancers 2014, 6, 796–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Thevenod, F. Expression and function of the lipocalin-2 (24p3/NGAL) receptor in rodent and human intestinal epithelia. PLoS ONE 2013, 8, e71586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauvois, B. NGAL-R expression and clinical features of CLL. INSERM UMRS1138, Paris, France. 2019; unpublished work. [Google Scholar]
- Cabedo Martinez, A.I.; Weinhaupl, K.; Lee, W.K.; Wolff, N.A.; Storch, B.; Zerko, S.; Konrat, R.; Kozminski, W.; Breuker, K.; Thevenod, F.; et al. Biochemical and Structural Characterization of the Interaction between the Siderocalin NGAL/LCN2 (Neutrophil Gelatinase-associated Lipocalin/Lipocalin 2) and the N-terminal Domain of Its Endocytic Receptor SLC22A17. J. Biol. Chem. 2016, 291, 2917–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Vitols, S.; Gruber, A.; Liliemark, J.; Wang, Y.; Liliemark, E. Etoposide-induced DNA strand breaks in relation to p-glycoprotein and topoisomerase II protein expression in leukaemic cells from patients with AML and CLL. Br. J. Haematol. 1999, 105, 420–427. [Google Scholar] [CrossRef]
- Rojas, R.; Roman, J.; Torres, A.; Ramirez, R.; Carracedo, J.; Lopez, R.; Garcia, J.M.; Martin, C.; Pintado, O. Inhibition of apoptotic cell death in B-CLL by interferon gamma correlates with clinical stage. Leukemia 1996, 10, 1782–1788. [Google Scholar]
- Bauvois, B. Effects of recombinant NGAL proteins on the survival of CLL cells. INSERM UMRS1138, Paris, France. 2016; unpublished work. [Google Scholar]
- Reed, J.C.; Kitada, S.; Kim, Y.; Byrd, J. Modulating apoptosis pathways in low-grade B-cell malignancies using biological response modifiers. Semin. Oncol. 2002, 29, 10–24. [Google Scholar] [CrossRef]
- Packham, G.; Stevenson, F.K. Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunology 2005, 114, 441–449. [Google Scholar] [CrossRef]
- Sun, X.M.; MacFarlane, M.; Zhuang, J.; Wolf, B.B.; Green, D.R.; Cohen, G.M. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 1999, 274, 5053–5060. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Haney, N.; Kropp, D.; Kabore, A.F.; Johnston, J.B.; Gibson, S.B. Lysophosphatidic acid (LPA) protects primary chronic lymphocytic leukemia cells from apoptosis through LPA receptor activation of the anti-apoptotic protein AKT/PKB. J. Biol. Chem. 2005, 280, 9498–9508. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.B.; Liu, Y.Q.; Cui, Y.F. Pathways to caspase activation. Cell. Biol. Int. 2005, 29, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Piedfer, M.; Dauzonne, D.; Tang, R.; N’Guyen, J.; Billard, C.; Bauvois, B. Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. FASEB J. 2011, 25, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.R.; Yang, Z.M. Regulation and function of signal transducer and activator of transcription 3. World J. Biol. Chem. 2014, 5, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Munoz, J.; Ugarte-Berzal, E.; Terol, M.J.; Van den Steen, P.E.; Hernandez del Cerro, M.; Roderfeld, M.; Roeb, E.; Opdenakker, G.; Garcia-Marco, J.A.; Garcia-Pardo, A. Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia B cell survival through its hemopexin domain. Cancer Cell 2010, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.C.; Pellegrini, S. The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell. Mol. Life Sci. 1999, 55, 1523–1534. [Google Scholar] [CrossRef]
- Severin, F.; Frezzato, F.; Visentin, A.; Martini, V.; Trimarco, V.; Carraro, S.; Tibaldi, E.; Brunati, A.M.; Piazza, F.; Semenzato, G.; et al. In Chronic Lymphocytic Leukemia the JAK2/STAT3 Pathway Is Constitutively Activated and Its Inhibition Leads to CLL Cell Death Unaffected by the Protective Bone Marrow Microenvironment. Cancers 2019, 11, 1939. [Google Scholar] [CrossRef] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Robak, E. Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert Opin. Investig. Drugs 2012, 21, 921–947. [Google Scholar] [CrossRef]
- Meydan, N.; Grunberger, T.; Dadi, H.; Shahar, M.; Arpaia, E.; Lapidot, Z.; Leeder, J.S.; Freedman, M.; Cohen, A.; Gazit, A.; et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 1996, 379, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Roli, L.; Pecoraro, V.; Trenti, T. Can NGAL be employed as prognostic and diagnostic biomarker in human cancers? A systematic review of current evidence. Int. J. Biol. Markers 2017, 32, e53–e61. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Maestro, R.; Polesel, J.; Catania, A.; Maira, F.; Signorelli, S.S.; McCubrey, J.A.; Libra, M. Roles of neutrophil gelatinase-associated lipocalin (NGAL) in human cancer. Oncotarget 2014, 5, 1576–1594. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Salvagno, G.L.; Banfi, G. Serum but not urine concentration of neutrophil gelatinase-associated lipocalin is influenced by acute leukocyte variations. Leuk. Lymphoma 2012, 53, 1643–1645. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Manshouri, T.; Estrov, Z.; Keating, M.J.; O’Brien, S.; Lerner, S.; Beran, M.; Kantarjian, H.M.; Freireich, E.J.; Albitar, M. High levels of vascular endothelial growth factor receptor-2 correlate with shortened survival in chronic lymphocytic leukemia. Clin. Cancer Res. 2001, 7, 795–799. [Google Scholar]
- Molica, S.; Vitelli, G.; Levato, D.; Gandolfo, G.M.; Liso, V. Increased serum levels of vascular endothelial growth factor predict risk of progression in early B-cell chronic lymphocytic leukaemia. Br. J. Haematol. 1999, 107, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B.; Dumont, J.; Mathiot, C.; Kolb, J.P. Production of matrix metalloproteinase-9 in early stage B-CLL: Suppression by interferons. Leukemia 2002, 16, 791–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foa, R.; Massaia, M.; Cardona, S.; Tos, A.G.; Bianchi, A.; Attisano, C.; Guarini, A.; di Celle, P.F.; Fierro, M.T. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: A possible regulatory role of TNF in the progression of the disease. Blood 1990, 76, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Baban, D.; Murray, J.; Earl, H.; Kerr, D.; Seymour, L. Quantitative analysis of vascular endothelial growth factor expression in chronic lymphocytic leukaemia. Int. J. Oncol. 1996, 8, 29–34. [Google Scholar] [CrossRef]
- Lee, Y.K.; Shanafelt, T.D.; Bone, N.D.; Strege, A.K.; Jelinek, D.F.; Kay, N.E. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: Implication for apoptosis resistance. Leukemia 2005, 19, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Gonzalez, A.; Aguilera-Montilla, N.; Ugarte-Berzal, E.; Bailon, E.; Cerro-Pardo, I.; Sanchez-Maroto, C.; Garcia-Campillo, L.; Garcia-Marco, J.A.; Garcia-Pardo, A. alpha4beta1 integrin associates with VEGFR2 in CLL cells and contributes to VEGF binding and intracellular signaling. Blood Adv. 2019, 3, 2144–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fan, Y.; Mei, Z. NGAL and NGALR overexpression in human hepatocellular carcinoma toward a molecular prognostic classification. Cancer Epidemiol. 2012, 36, e294–e299. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.P.; Lv, Z.; Wu, B.L.; Wu, Z.Y.; Shen, J.H.; Wu, J.Y.; Xu, X.E.; Huang, Q.; Shen, J.; Chen, H.B.; et al. Neutrophil gelatinase-associated lipocalin and its receptor: Independent prognostic factors of oesophageal squamous cell carcinoma. J. Clin. Pathol. 2011, 64, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.F.; Jin, T.; Shen, J.H.; Shen, Z.Y.; Zheng, Z.C.; Zhang, Z.L.; Xu, L.Y.; Li, E.M.; Xu, H.X. NGAL and NGALR are frequently overexpressed in human gliomas and are associated with clinical prognosis. J. Neuro Oncol. 2011, 104, 119–127. [Google Scholar] [CrossRef]
- Liu, F.; Li, N.; Yang, W.; Wang, R.; Yu, J.; Wang, X. The expression analysis of NGAL and NGALR in clear cell renal cell carcinoma. Gene 2018, 676, 269–278. [Google Scholar] [CrossRef]
- Lv, Z.; Xu, L.Y.; Shen, Z.Y.; Zhang, F.R.; Xu, X.E.; Li, E.M. Overexpression of neutrophil gelatinase-associated lipocalin and its receptor in colorectal carcinoma: Significant correlation with cell differentiation and tumour invasion. Oncol. Lett. 2010, 1, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C.; Li, S.J.; Chen, Z.L.; Li, G.; Zhang, Q.; Li, D.J. Expression and Prognosis Analyses of Runt-Related Transcription Factor Family in Human Leukemia. Mol. Ther. Oncolytics 2019, 12, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Wang, S.Z.; Green, M.R. Transcription and signalling pathways involved in BCR-ABL-mediated misregulation of 24p3 and 24p3R. EMBO J. 2009, 28, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Levanon, D.; Groner, Y. Structure and regulated expression of mammalian RUNX genes. Oncogene 2004, 23, 4211–4219. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Tsunematsu, T.; Takata, T. Oncogenic role of RUNX3 in head and neck cancer. J. Cell. Biochem. 2011, 112, 387–393. [Google Scholar] [CrossRef]
- Lagneaux, L.; Delforge, A.; Bron, D.; De Bruyn, C.; Stryckmans, P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood 1998, 91, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825. [Google Scholar] [CrossRef] [Green Version]
- Forte, D.; Krause, D.S.; Andreeff, M.; Bonnet, D.; Mendez-Ferrer, S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica 2019, 104, 1928–1934. [Google Scholar] [CrossRef]
- Van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Purroy, N.; Wu, C.J. Coevolution of Leukemia and Host Immune Cells in Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimarco, V.; Ave, E.; Facco, M.; Chiodin, G.; Frezzato, F.; Martini, V.; Gattazzo, C.; Lessi, F.; Giorgi, C.A.; Visentin, A.; et al. Cross-talk between chronic lymphocytic leukemia (CLL) tumor B cells and mesenchymal stromal cells (MSCs): Implications for neoplastic cell survival. Oncotarget 2015, 6, 42130–42149. [Google Scholar] [CrossRef] [PubMed]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef]
- Pepper, C.; Lin, T.T.; Pratt, G.; Hewamana, S.; Brennan, P.; Hiller, L.; Hills, R.; Ward, R.; Starczynski, J.; Austen, B.; et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood 2008, 112, 3807–3817. [Google Scholar] [CrossRef] [PubMed]
- Awan, F.T.; Kay, N.E.; Davis, M.E.; Wu, W.; Geyer, S.M.; Leung, N.; Jelinek, D.F.; Tschumper, R.C.; Secreto, C.R.; Lin, T.S.; et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood 2009, 113, 535–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobessi, S.; Laurenti, L.; Longo, P.G.; Carsetti, L.; Berno, V.; Sica, S.; Leone, G.; Efremov, D.G. Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia 2009, 23, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Wu, J.Y.; Harris, D.M.; Liu, Z.; Li, P.; Hazan-Halevy, I.; Ferrajoli, A.; Burger, J.A.; O’Brien, S.; Jain, N.; et al. Stimulation of the B-cell receptor activates the JAK2/STAT3 signaling pathway in chronic lymphocytic leukemia cells. Blood 2014, 123, 3797–3802. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.; Rowland, T.A.; Sreekantham, B.; Godbersen, C.; Best, S.R.; Kaur, P.; Loriaux, M.M.; Spurgeon, S.E.F.; Danilova, O.V.; Danilov, A.V. SYK inhibition thwarts the BAFF—B-cell receptor crosstalk and thereby antagonizes Mcl-1 in chronic lymphocytic leukemia. Haematologica 2017, 102, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Qing, Y.; Stark, G.R. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J. Biol. Chem. 2004, 279, 41679–41685. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.; Sher, D.A.; Alsayed, Y.; Pons, S.; Colamonici, O.R.; Fish, E.N.; White, M.F.; Platanias, L.C. Interaction of p59fyn with interferon-activated Jak kinases. Biochem. Biophys. Res. Commun. 1997, 235, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.J.; Holtzman, M.J.; Chen, C.C. Differential role of Janus family kinases (JAKs) in interferon-gamma-induced lung epithelial ICAM-1 expression: Involving protein interactions between JAKs, phospholipase Cgamma, c-Src, and STAT1. Mol. Pharmacol. 2004, 65, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Becker, T.M.; Boyd, S.C.; Mijatov, B.; Gowrishankar, K.; Snoyman, S.; Pupo, G.M.; Scolyer, R.A.; Mann, G.J.; Kefford, R.F.; Zhang, X.D.; et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene 2014, 33, 1158–1166. [Google Scholar] [CrossRef] [Green Version]
- Contri, A.; Brunati, A.M.; Trentin, L.; Cabrelle, A.; Miorin, M.; Cesaro, L.; Pinna, L.A.; Zambello, R.; Semenzato, G.; Donella-Deana, A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J. Clin. Investig. 2005, 115, 369–378. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Fedorchenko, O.; Rosen, N.; Koch, M.; Barthel, R.; Winarski, T.; Florin, A.; Wunderlich, F.T.; Reinart, N.; Hallek, M. LYN Kinase in the Tumor Microenvironment Is Essential for the Progression of Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, D.J.; Scharenberg, A.M.; Park, H.; Wahl, M.I.; Lin, S.; Kato, R.M.; Fluckiger, A.C.; Witte, O.N.; Kinet, J.P. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 1996, 271, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.; Ozer, Z.; Vassilev, A. Bruton’s tyrosine kinase prevents activation of the anti-apoptotic transcription factor STAT3 and promotes apoptosis in neoplastic B-cells and B-cell precursors exposed to oxidative stress. Br. J. Haematol. 2007, 136, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Wu, C.L.; Ping, S.Y.; Huang, Y.L.; Shen, K.H. NGAL can alternately mediate sunitinib resistance in renal cell carcinoma. J. Urol. 2014, 192, 559–566. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Bergui, L.; Bonello, L.; Horenstein, A.L.; Tamagnone, L.; Boumsell, L.; Malavasi, F. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood 2005, 105, 3042–3050. [Google Scholar] [CrossRef]
- Zucchetto, A.; Vaisitti, T.; Benedetti, D.; Tissino, E.; Bertagnolo, V.; Rossi, D.; Bomben, R.; Dal Bo, M.; Del Principe, M.I.; Gorgone, A.; et al. The CD49d/CD29 complex is physically and functionally associated with CD38 in B-cell chronic lymphocytic leukemia cells. Leukemia 2012, 26, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. Molecular Mode of Action of TRAIL Receptor Agonists-Common Principles and Their Translational Exploitation. Cancers 2019, 11, 954. [Google Scholar] [CrossRef] [Green Version]
- Bolignano, D.; Lacquaniti, A.; Coppolino, G.; Donato, V.; Campo, S.; Fazio, M.R.; Nicocia, G.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Papassotiriou, G.P.; Kastritis, E.; Gkotzamanidou, M.; Christoulas, D.; Eleutherakis-Papaiakovou, E.; Migkou, M.; Gavriatopoulou, M.; Roussou, M.; Margeli, A.; Papassotiriou, I.; et al. Neutrophil Gelatinase--Associated Lipocalin and Cystatin C Are Sensitive Markers of Renal Injury in Patients With Multiple Myeloma. Clin. Lymphoma Myeloma Leukemia 2016, 16, 29–35. [Google Scholar] [CrossRef]
- Di Carlo, A. Evaluation of neutrophil gelatinase-associated lipocalin (NGAL), matrix metalloproteinase-9 (MMP-9) and their complex MMP-9/NGAL in sera and urine of patients with kidney tumors. Oncol. Lett. 2013, 5, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.K.; Xu, L.Y.; Lu, X.F.; Liao, L.D.; Cai, W.J.; Shen, Z.Y.; Li, E.M. A novel alternative spliced variant of neutrophil gelatinase-associated lipocalin receptor in oesophageal carcinoma cells. Biochem. J. 2007, 403, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, P.S.; Bayne, R.A.; Robinson, L.L.; Fulton, N.; Anderson, R.A. Developmental changes in expression of myeloid cell leukemia-1 in human germ cells during oogenesis and early folliculogenesis. J. Clin. Endocrinol. Metab. 2002, 87, 3417–3427. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.J.; Huang, Y.H.; Au, H.K.; Wang, L.M.; Chu, S.T. The cancer marker neutrophil gelatinase-associated lipocalin is highly expressed in human endometrial hyperplasia. Mol. Biol. Rep. 2012, 39, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.X.; Yu, R.T.; Liu, Z. Inhibition of two gastric cancer cell lines induced by fucoxanthin involves downregulation of Mcl-1 and STAT3. Hum. Cell 2018, 31, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Sancéau, J.; Boyd, D.D.; Seiki, M.; Bauvois, B. Interferons inhibit tumor necrosis factor-alpha-mediated matrix metalloproteinase-9 activation via interferon regulatory factor-1 binding competition with NF-kappa B. J. Biol. Chem. 2002, 277, 35766–35775. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Dohner, H.; Hillmen, P.; Keating, M.J.; Montserrat, E.; Rai, K.R.; et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008, 111, 5446–5456. [Google Scholar] [CrossRef] [Green Version]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Furstenau, M.; Hallek, M.; Eichhorst, B. Sequential and combination treatments with novel agents in chronic lymphocytic leukemia. Haematologica 2019, 104, 2144–2154. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Wagner, C.; de Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keseru, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Dai, N.; Tang, Z.; Fang, H. Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur. J. Med. Chem. 2018, 146, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. MCL-1 inhibitors—Where are we now (2019)? Expert Opin. Ther. Pat. 2019, 29, 909–919. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex/Age | Stage | IGHV Mutation | FISH | Karyotype | Therapy |
|---|---|---|---|---|---|---|
| 1 | M/71 | A | M | Trisomy 12 | 1 abnormality | None |
| 2 | M/63 | A | ND | 13q- | Complex | None |
| 3 | M/90 | A | M | ND | ND | None |
| 4 | M/88 | A | M | 13q- | Normal | None |
| 5 | M/76 | A | ND | 13q- | Normal | None |
| 6 | M/87 | C | M | 17p-, 13q- | Normal | None |
| 7 | M/66 | B | M | 13q- | 2 abnormalities | None |
| 8 | M/71 | A | ND | 13q- | Normal | None |
| 9 | M/71 | A | M | 11q-, 13q- | 1 abnormality | None |
| 10 | F/69 | B | ND | 13q- | Normal | None |
| 11 | F/74 | A | UM | 13q- | Normal | None |
| 12 | F/74 | A | M | Normal | Normal | None |
| 13 | F/78 | A | M | 13q- | Normal | None |
| 14 | M/73 | A | M | Normal | Normal | None |
| 15 | F/68 | A | ND | 13q- | 1 abnormality | None |
| 16 | F/62 | B | UM | 13q- | Normal | None |
| 17 | F/69 | A | M | 13q- | Normal | None |
| 18 | F/87 | A | ND | 13q- | Normal | None |
| 19 | F/83 | A | ND | Normal | Normal | None |
| 20 | M/86 | A | ND | Normal | Normal | None |
| 21 | M/86 | A | M | Trisomy 12 | Complex | None |
| 22 | M/82 | A | ND | 11q-, 13q- | Normal | None |
| 23 | F/77 | A | UM | 13q- | Failure | None |
| 24 | M/74 | A | ND | 13q- | 2 abnormalities | None |
| 25 | M/86 | A | M | 13q- | ND | None |
| 26 | F/76 | A | UM | 11q-, 13q- | Normal | None |
| 27 | F/88 | A | ND | ND | ND | None |
| 28 | F/64 | A | M | 13q- | Normal | None |
| 29 | M/70 | A | ND | 13q- | Normal | None |
| 30 | M/75 | A | M | 13q- | Normal | None |
| 31 | F/67 | A | ND | 13q- | Normal | None |
| 32 | F/74 | A | ND | 13q- | 1 abnormality | None |
| 33 | M/76 | A | ND | 13q- | Normal | None |
| 34 | M/68 | B | ND | 11q- | Complex | None |
| 35 | F/85 | A | M | 13q- | Normal | None |
| 36 | F/70 | A | ND | 13q- | Normal | None |
| 37 | F/52 | A | ND | 13q- | Normal | None |
| 38 | F/79 | A | ND | 13q- | Normal | None |
| 39 | F/54 | B | ND | Trisomy 12 | 1 abnormality | None |
| 40 | M/76 | A | M | 13q- | Normal | None |
| 41 | F/57 | A | ND | 13q- | Normal | None |
| 42 | M/72 | A | M | 13q- | ND | None |
| 43 | F/84 | A | ND | Normal | Normal | None |
| 44 | F/80 | B | ND | 11q- | 1 abnormality | None |
| 45 | M/62 | A | ND | Normal | Normal | None |
| 46 | M/75 | B | UM | Trisomy 12 | 1 abnormality | None |
| 47 | F/73 | A | ND | 13q- | Failure | None |
| 48 | M/59 | A | ND | ND | ND | None |
| 49 | F/75 | A | ND | 13q- | Normal | None |
| 50 | M/61 | B | ND | 13q-, Trisomy 12 | Complex | None |
| 51 | F/82 | A | ND | 13q- | Normal | None |
| 52 | F/53 | A | UM | 13q- | 2 abnormalities | None |
| 53 | M/71 | A | UM | 11q-, 13q- | ND | None |
| 54 | F/96 | A | UM | Trisomy 12 | 1 abnormality | None |
| 55 | M/72 | A | ND | Normal | Normal | None |
| 56 | F/85 | A | ND | Normal | 2 abnormalities | None |
| 57 | F/69 | A | M | Normal | Normal | None |
| 58 | F/51 | B | UM | 11q-, 13q- | 1 abnormality | None |
| 59 | M/63 | B | UM | 13q- | Normal | None |
| 60 | M/70 | B | UM | 11q-, 13q- | 2 abnormalities | None |
| 61 | F/80 | B | ND | 11q- | 1 abnormality | Ibrutinib/relapse |
| 62 | M/62 | B | UM | 11q-, 13q- | 1 abnormality | FCR/relapse |
| 63 | F/78 | A | M | 13q- | 1 abnormality | RCb/relapse |
| 64 | M/54 | B | UM | 13q- | 2 abnormalities | Ibrutinib/remission |
| 65 | F/78 | C | UM | 17p-, 13q-, Trisomy 12 | Complex | Ibrutinib/remission |
| 66 | M/76 | B | ND | 17p-, 13q- | 2 abnormalities | Ibrutinib/relapse |
| 67 | M/80 | B | UM | 17p-, 13q- | Complex | Ibrutinib/relapse |
| 68 | M/54 | C | UM | 17p-, 11q- | Complex | Ibrutinib/remission |
| 69 | F/72 | B | UM | 17p-, Trisomy 12 | Complex | Ibrutinib/remission |
| 70 | M/92 | A | ND | ND | ND | Cb/relapse |
| 71 | M/66 | C | UM | 13q-, Trisomy 12 | Complex | Ibrutinib/remission |
| 72 | M/70 | A | M&UM | 11q- | Failure | FCR/relapse |
| 73 | F/77 | A | M | Normal | ND | FC/relapse |
| 74 | M/88 | C | UM | 17p-, Trisomy 12 | Normal | RCb/remission |
| 75 | M/82 | C | UM | 11q-, 13q- | Complex | BR/relapse |
| 76 | M/79 | B | UM | Normal | Normal | BR/remission |
| 77 | M/64 | A | UM | Trisomy 12 | 1 abnormality | FCR/cyclosporine/remission |
| 78 | F/75 | B | M | 13q-, Trisomy 12 | 1 abnormality | FCR/remission |
| 79 | F/84 | B | ND | Normal | Complex | Ibrutinib/remission |
| 80 | F/64 | B | UM | 13q- | Normal | FCR/remission |
| 81 | M/69 | B | ND | 11q- | Complex | FCR x4/remission |
| 82 | M/75 | B | M | Trisomy 12 | 1 abnormality | BR/remission |
| 83 | M/74 | C | M | 11q-, 13q- | ND | MiniCHOP/remission |
| 84 | M/81 | C | M | 11q-, 13q- | ND | Alemtuzumab/relapse |
| 85 | M/75 | B | UM | 17p-, 13q- | 2 abnormalities | FCR x2/relapse |
| Patient P16/Months | Lymphocyte Count (G/L) | Neutrophil Count (G/L) |
| 0 | 159.6 | 12.55 |
| 5 | 179.86 | 26 |
| 7 | 189.38 | 12.91 |
| 9 | 196.14 | 2.13 |
| 11 | 239.94 | 4.0 |
| 12 | 248.5 | 5.29 |
| 19 (treated) | 0.26 | 0.80 |
| 27 (treated) | 0.59 | 2.28 |
| Correlation with: | p-value | p-value |
| serum NGAL | 0.0011 | 0.6191 |
| serum CPX | 0.0011 | 0.6191 |
| Patient P46/Months | Lymphocyte Count (G/L) | Neutrophil Count (G/L) |
| 0 | 146 | 1.5 |
| 1.5 | 184 | 3.9 |
| 4 | 219.5 | 2.3 |
| 10 (treated) | 1.11 | 4.39 |
| Correlation with: | p-value | p-value |
| serum NGAL | 0.0833 | 0.7500 |
| serum CPX | 0.3333 | 0.3333 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauvois, B.; Pramil, E.; Jondreville, L.; Chapiro, E.; Quiney, C.; Maloum, K.; Susin, S.A.; Nguyen-Khac, F. Relation of Neutrophil Gelatinase-Associated Lipocalin Overexpression to the Resistance to Apoptosis of Tumor B Cells in Chronic Lymphocytic Leukemia. Cancers 2020, 12, 2124. https://doi.org/10.3390/cancers12082124
Bauvois B, Pramil E, Jondreville L, Chapiro E, Quiney C, Maloum K, Susin SA, Nguyen-Khac F. Relation of Neutrophil Gelatinase-Associated Lipocalin Overexpression to the Resistance to Apoptosis of Tumor B Cells in Chronic Lymphocytic Leukemia. Cancers. 2020; 12(8):2124. https://doi.org/10.3390/cancers12082124
Chicago/Turabian StyleBauvois, Brigitte, Elodie Pramil, Ludovic Jondreville, Elise Chapiro, Claire Quiney, Karim Maloum, Santos A. Susin, and Florence Nguyen-Khac. 2020. "Relation of Neutrophil Gelatinase-Associated Lipocalin Overexpression to the Resistance to Apoptosis of Tumor B Cells in Chronic Lymphocytic Leukemia" Cancers 12, no. 8: 2124. https://doi.org/10.3390/cancers12082124