Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Methods
2.1. Clinical and Mutational Database
2.2. Statistical Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cui, R.R.; Wright, J.D.; Hou, J.Y. Uterine leiomyosarcoma: A review of recent advances in molecular biology, clinical management and outcome. BJOG Int. J. Obstet. Gynaecol. 2017, 124, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.E.; Aynardi, J.T.; Chu, C.S. Uterine leiomyosarcoma: A review of the literature and update on management options. Gynecol. Oncol. 2018, 151, 562–572. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Serrano, C.; Hensley, M.L.; Ray-Coquard, I. Soft tissue and uterine leiomyosarcoma. J. Clin. Oncol. 2018, 36, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Hosh, M.; Antar, S.; Nazzal, A.; Warda, M.; Gibreel, A.; Refky, B. Uterine Sarcoma: Analysis of 13,089 Cases Based on Surveillance, Epidemiology, and End Results Database. Int. J. Gynecol. Cancer 2016, 26, 1098–1104. [Google Scholar] [CrossRef]
- Wu, T.I.; Chang, T.C.; Hsueh, S.; Hsu, K.H.; Chou, H.H.; Huang, H.J.; Lai, C.H. Prognostic factors and impact of adjuvant chemotherapy for uterine leiomyosarcoma. Gynecol. Oncol. 2006, 100, 166–172. [Google Scholar] [CrossRef]
- Kapp, D.S.; Shin, J.Y.; Chan, J.K. Prognostic factors and survival in 1396 patients with uterine leiomyosarcomas: Emphasis on impact of lymphadenectomy and oophorectomy. Cancer 2008, 112, 820–830. [Google Scholar] [CrossRef]
- Friedman, C.F.; Hensley, M.L. Options for Adjuvant Therapy for Uterine Leiomyosarcoma. Curr. Treat. Opt. Oncol. 2018, 19, e7. [Google Scholar] [CrossRef]
- Rizzo, A.; Pantaleo, M.A.; Saponara, M.; Nannini, M. Current status of the adjuvant therapy in uterine sarcoma: A literature review. World J. Clin. Cases 2019, 7, 1753–1763. [Google Scholar] [CrossRef]
- Casali, P.; Abecassis, N.; Aro, H.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Soft tissue and visceral sarcomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv51–iv67. [Google Scholar] [CrossRef]
- D’Ambrosio, L.; Touati, N.; Blay, J.Y.; Grignani, G.; Flippot, R.; Czarnecka, A.M.; Piperno-Neumann, S.; Martin-Broto, J.; Sanfilippo, R.; Katz, D.; et al. European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, or doxorubicin alone as a first-line treatment for advanced leiomyosarcoma: A propensity score matching analysis from the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Cancer 2020, 126, 2637–2647. [Google Scholar]
- Vincenzi, B.; Stacchiotti, S.; Collini, P.; Pantano, F.; Rabitti, C.; Perrone, G.; Iuliani, M.; Baldi, A.; Badalamenti, G.; Sanfilippo, R.; et al. Human equilibrative nucleoside transporter 1 gene expression is associated with gemcitabine efficacy in advanced leiomyosarcoma and angiosarcoma. Br. J. Cancer 2017, 117, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Arend, R.C.; Toboni, M.D.; Montgomery, A.M.; Burger, R.A.; Olawaiye, A.B.; Monk, B.J.; Herzog, T.J. Systemic Treatment of Metastatic/Recurrent Uterine Leiomyosarcoma: A Changing Paradigm. Oncologist 2018, 23, 1533–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, N.; Aavikko, M.; Heikkinen, T.; Taipale, M.; Taipale, J.; Koivisto-Korander, R.; Bützow, R.; Vahteristo, P. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016, 12, e1005850. [Google Scholar]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [Green Version]
- Cuppens, T.; Moisse, M.; Depreeuw, J.; Annibali, D.; Colas, E.; Gil-Moreno, A.; Huvila, J.; Carpén, O.; Zikán, M.; Matias-Guiu, X.; et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018, 142, 1230–1243. [Google Scholar] [CrossRef] [Green Version]
- Tsuyoshi, H.; Yoshida, Y. Molecular biomarkers for uterine leiomyosarcoma and endometrial stromal sarcoma. Cancer Sci. 2018, 109, 1743–1752. [Google Scholar] [CrossRef]
- Hemming, M.L.; Klega, K.S.; Rhoades, J.; Ha, G.; Acker, K.E.; Andersen, J.L.; Thai, E.; Nag, A.; Thorner, A.R.; Raut, C.P.; et al. Detection of Circulating Tumor DNA in Patients with Leiomyosarcoma with Progressive Disease. Jco. Precis. Oncol. 2019, 2019. [Google Scholar] [CrossRef]
- Hensley, M.L.; Chavan, S.S.; Solit, D.B.; Murali, R.; Soslow, R.; Chiang, S.; Jungbluth, A.A.; Bandlamudi, C.; Srinivasan, P.; Tap, W.D.; et al. Genomic Landscape of Uterine Sarcomas Defined through Prospective Clinical Sequencing. Clin. Cancer Res. 2020, 26, 3881–3888. [Google Scholar] [CrossRef] [Green Version]
- Croce, S.; Lesluyes, T.; Valle, C.; M’Hamdi, L.; Thébault, N.; Pérot, G.; Stoeckle, E.; Noël, J.-C.; Fontanges, Q.; Devouassoux-Shisheboran, M.; et al. The Nanocind Signature Is an Independent Prognosticator of Recurrence and Death in Uterine Leiomyosarcomas. Clin. Cancer Res. 2020, 26, 855–861. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Akso, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, e7538. [Google Scholar] [CrossRef]
- Koschmann, C.; Calinescu, A.A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, e144. [Google Scholar] [CrossRef] [Green Version]
- Oppel, F.; Tao, T.; Shi, H.; Ross, K.N.; Zimmerman, M.W.; He, S.; Tong, G.; Aster, J.C.; Look, T.A. Loss of ATRX cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 2019, 15, e1008039. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; D’Ambrosio, L.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Stacchiotti, S.; Bertulli, R.; Piana, R.; et al. Trabectedin and Olaparib in patients with advanced and non-resectable bone and soft-tissue sarcomas (TOMAS): An open-label, phase 1b study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371. [Google Scholar] [CrossRef]
- Seligson, N.D.; Kautto, E.A.; Passen, E.N.; Stets, C.; Toland, A.E.; Millis, S.Z.; Meyer, C.F.; Hays, J.L.; Chen, J.L. BRCA1/2 Functional Loss Defines a Targetable Subset in Leiomyosarcoma. Oncologist 2019, 24, 973–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 2011, 334, 252–255. [Google Scholar] [CrossRef]
- Koh, W.J.; Abu-Rustum, N.R.; Bean, S.; Bradley, K.; Campos, S.M.; Cho, K.R.; Chon, H.S.; Chu, C.; Cohn, D.; Crispens, M.A.; et al. Uterine Neoplasms, Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2018, 16, 170–199. [Google Scholar] [CrossRef] [Green Version]
- Le Cesne, A.; Cresta, S.; Maki, R.G.; Blay, J.Y.; Verweij, J.; Poveda, A.; Casali, P.G.; Balaña, C.; Schöffski, P.; Grosso, F.; et al. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer 2012, 48, 3036–3044. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Subgroup | Class | Average OS (Months) | SE | Log-Rank p-Value |
|---|---|---|---|---|
| All | TP53 wild-type | 117.9 | 20.5 | 0.18 |
| TP53 mutant | 92.1 | 12.7 | ||
| RB1 wild-type | 89.2 | 12.9 | 0.22 | |
| RB1 mutant | 131.4 | 17.7 | ||
| ATRX wild-type | 111.4 | 15.1 | 0.63 | |
| ATRX mutant | 101.9 | 15.9 | ||
| PTEN wild-type | 93.3 | 15.1 | 0.52 | |
| PTEN mutant | 146.9 | 15.9 | ||
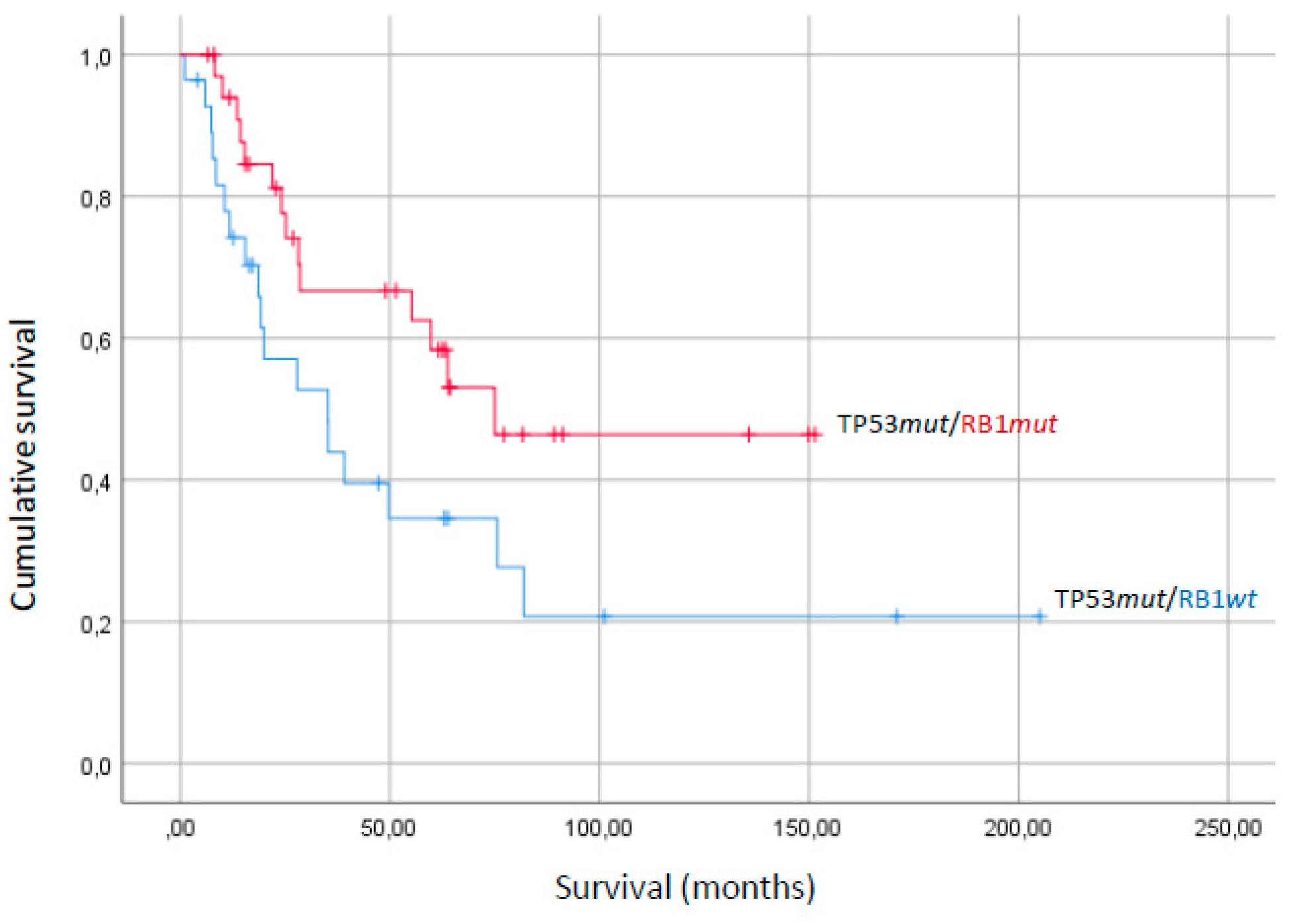
| TP53 mutant | RB1 wild-type | 67.1 | 16.4 | 0.04 |
| RB1 mutant | 89.9 | 11.8 | ||
| RB1 wild-type | TP53 wild-type | 99.2 | 14.4 | 0.05 |
| TP53 mutant | 67.1 | 16.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astolfi, A.; Nannini, M.; Indio, V.; Schipani, A.; Rizzo, A.; Perrone, A.M.; De Iaco, P.; Pirini, M.G.; De Leo, A.; Urbini, M.; et al. Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers 2020, 12, 2126. https://doi.org/10.3390/cancers12082126
Astolfi A, Nannini M, Indio V, Schipani A, Rizzo A, Perrone AM, De Iaco P, Pirini MG, De Leo A, Urbini M, et al. Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers. 2020; 12(8):2126. https://doi.org/10.3390/cancers12082126
Chicago/Turabian StyleAstolfi, Annalisa, Margherita Nannini, Valentina Indio, Angela Schipani, Alessandro Rizzo, Anna Myriam Perrone, Pierandrea De Iaco, Maria Giulia Pirini, Antonio De Leo, Milena Urbini, and et al. 2020. "Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile" Cancers 12, no. 8: 2126. https://doi.org/10.3390/cancers12082126
APA StyleAstolfi, A., Nannini, M., Indio, V., Schipani, A., Rizzo, A., Perrone, A. M., De Iaco, P., Pirini, M. G., De Leo, A., Urbini, M., Secchiero, P., & Pantaleo, M. A. (2020). Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers, 12(8), 2126. https://doi.org/10.3390/cancers12082126