The Curcumin Analogue, EF-24, Triggers p38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells

 ,
,  and
and 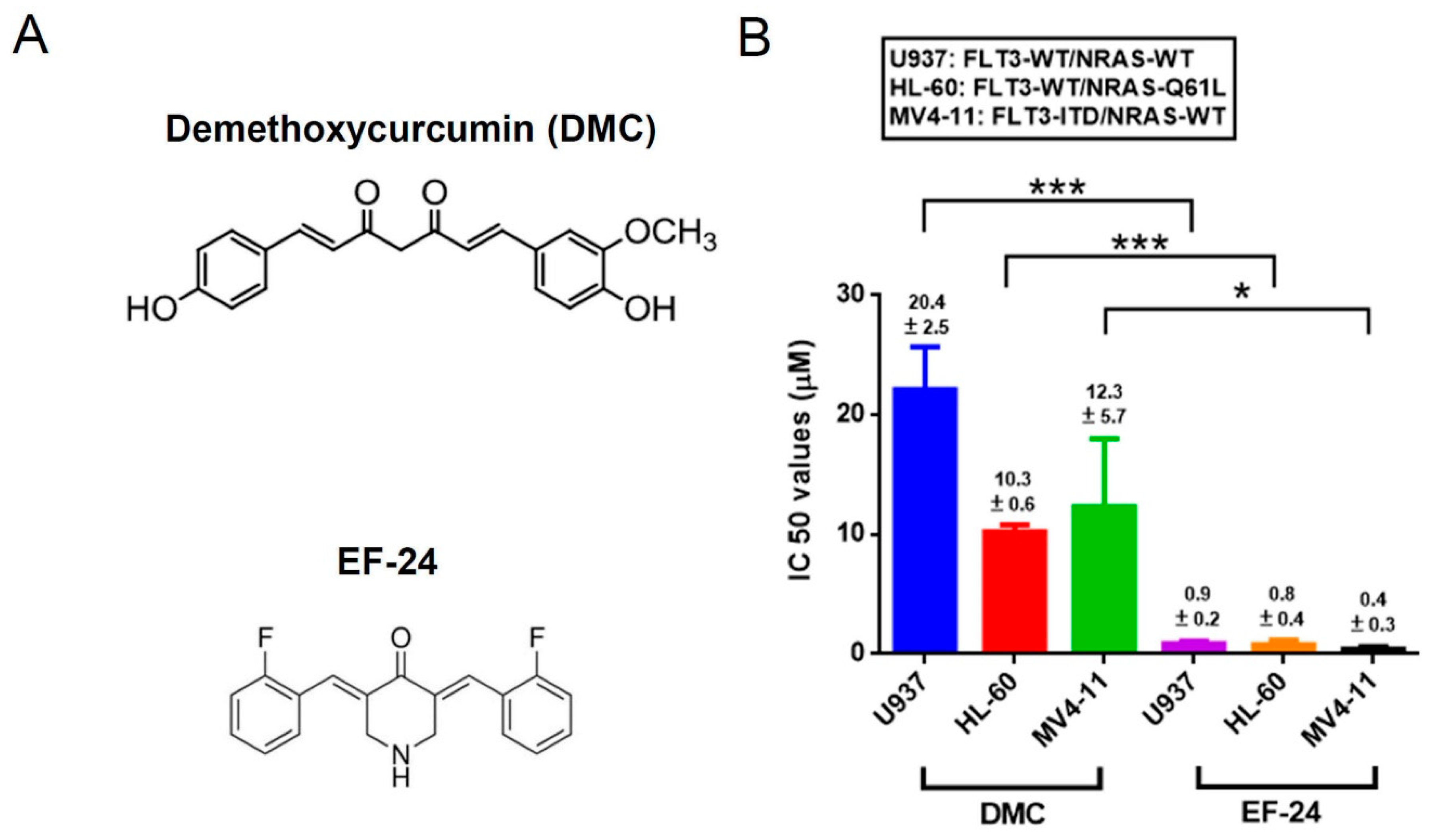{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
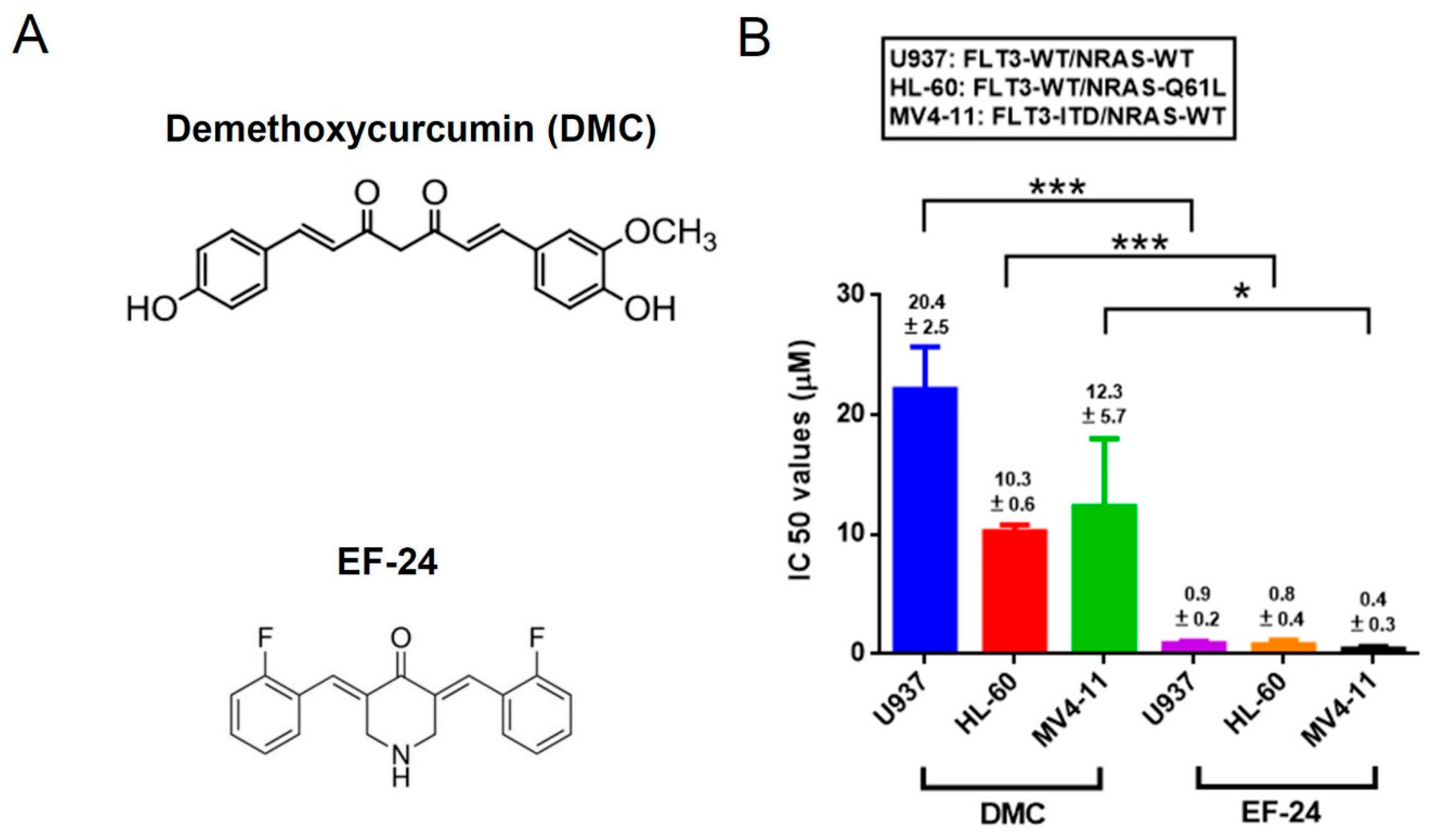
2.1. EF-24 Exerts a More Potent Effect than DMC in Decreasing the Proportion of Viable Cells in AML Cell Lines Harboring Different FLT3 and NRAS Statuses
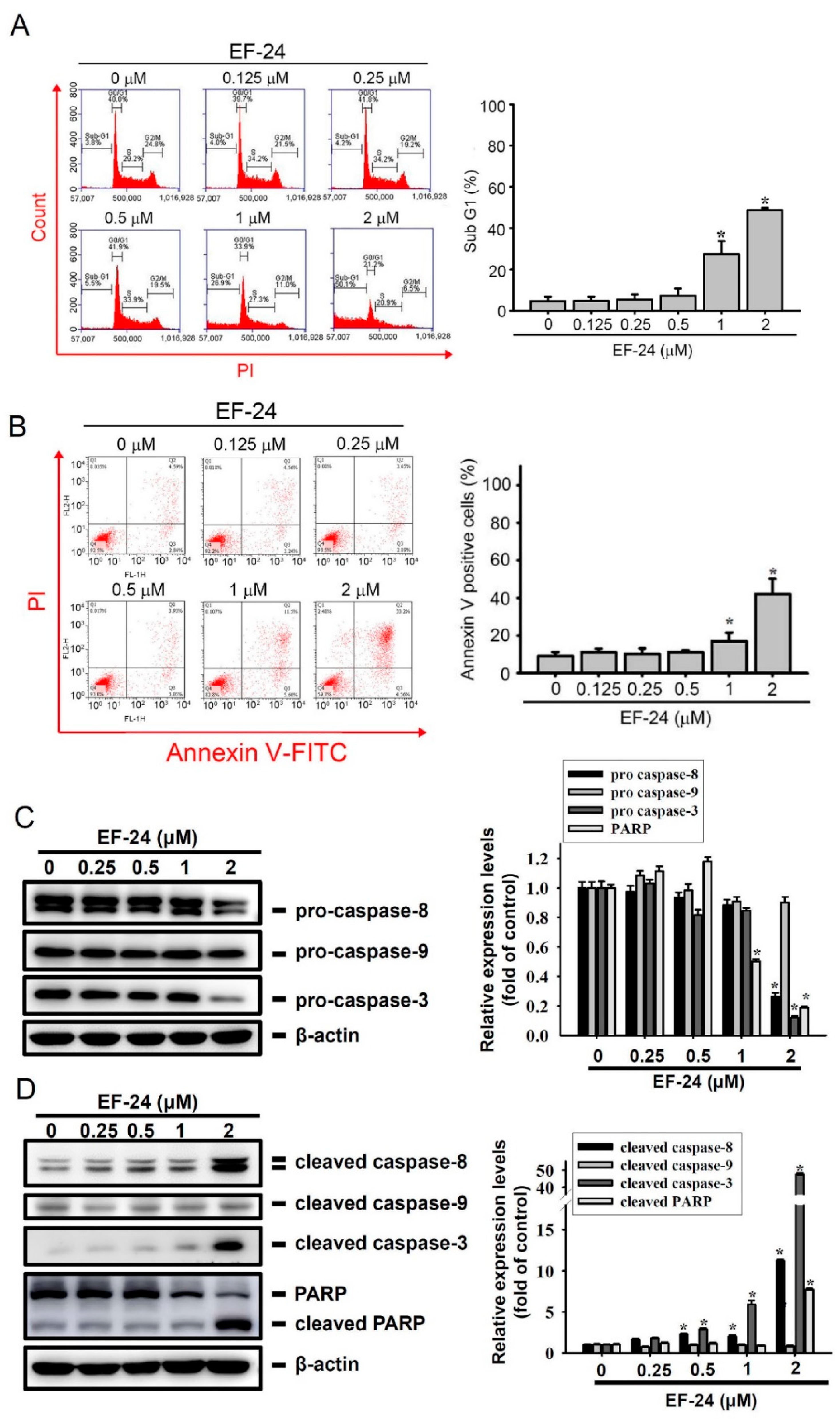
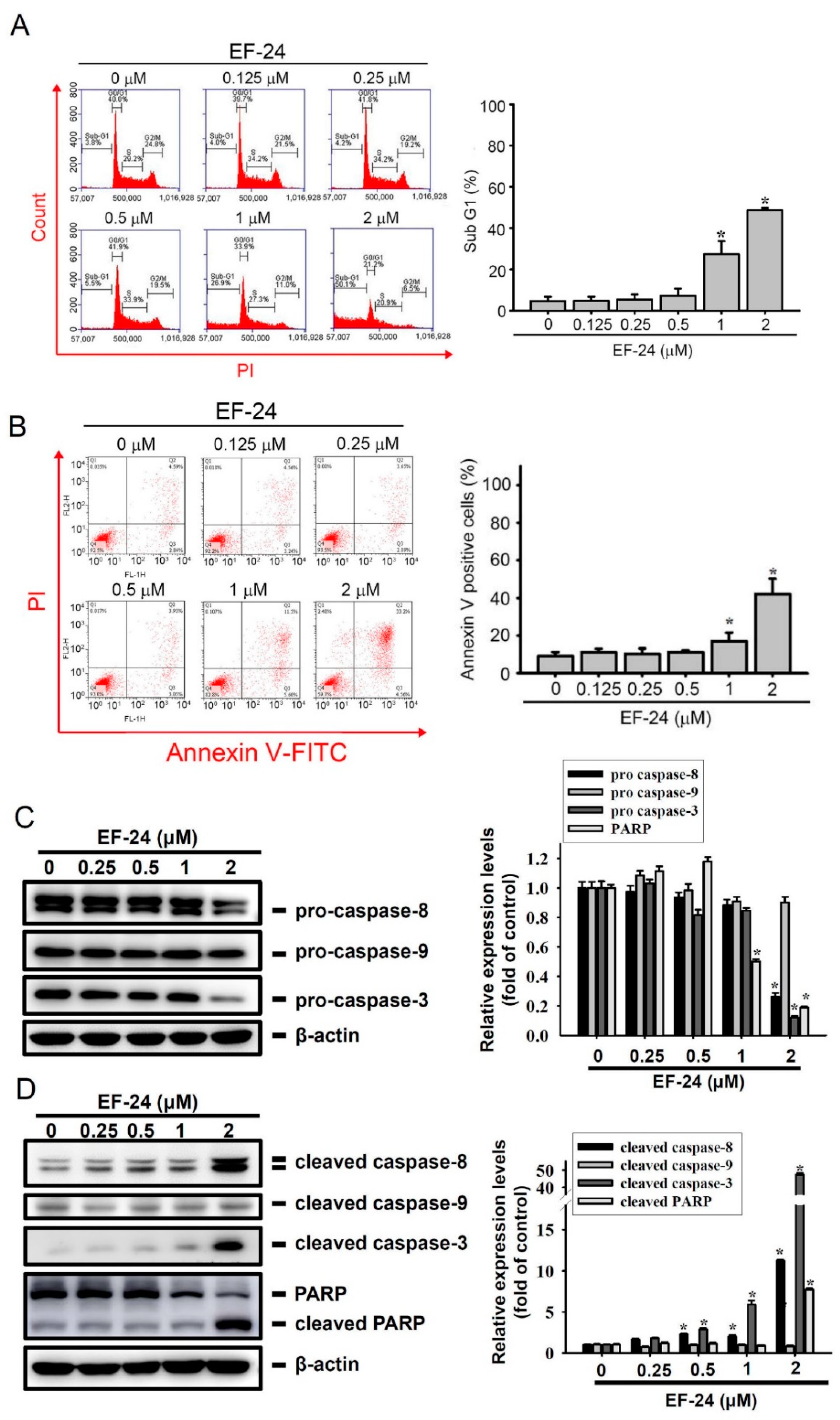
2.2. EF-24 Treatment Results in Extrinsic Apoptotic Cell Death of AML Cells
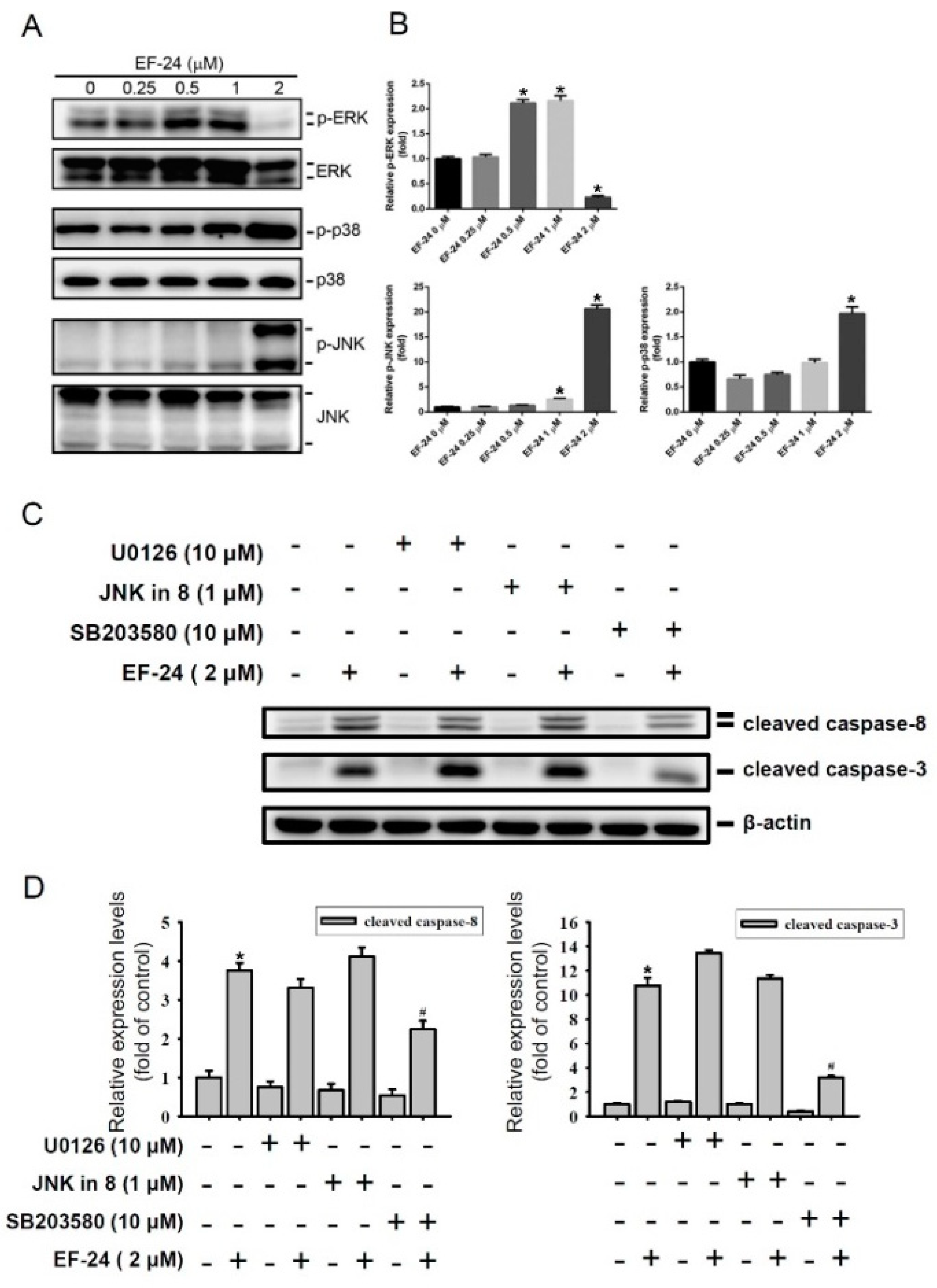
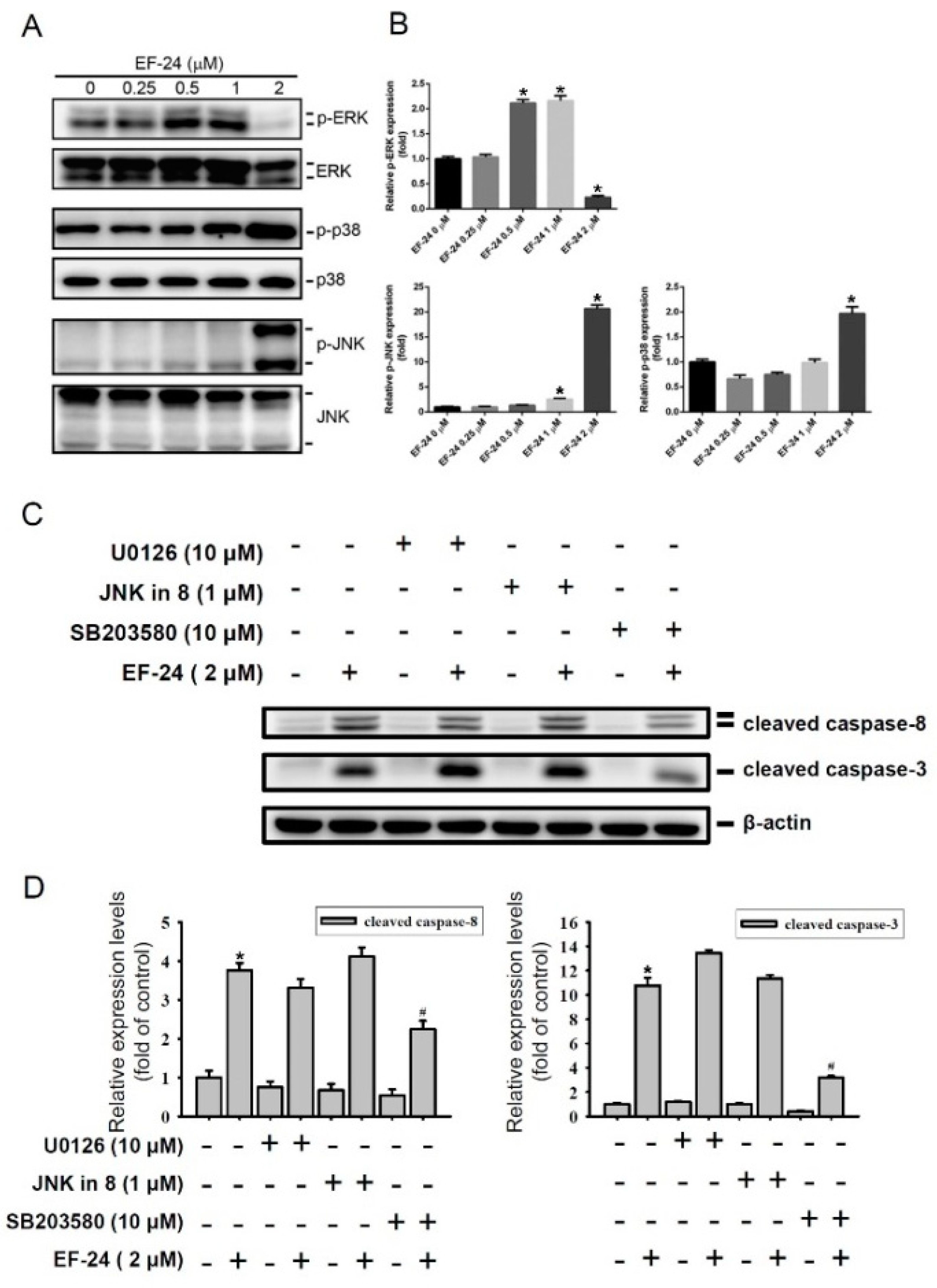
2.3. EF-24-Induced Apoptosis of HL-60 AML Cells via Triggering p38 MAPK Activation
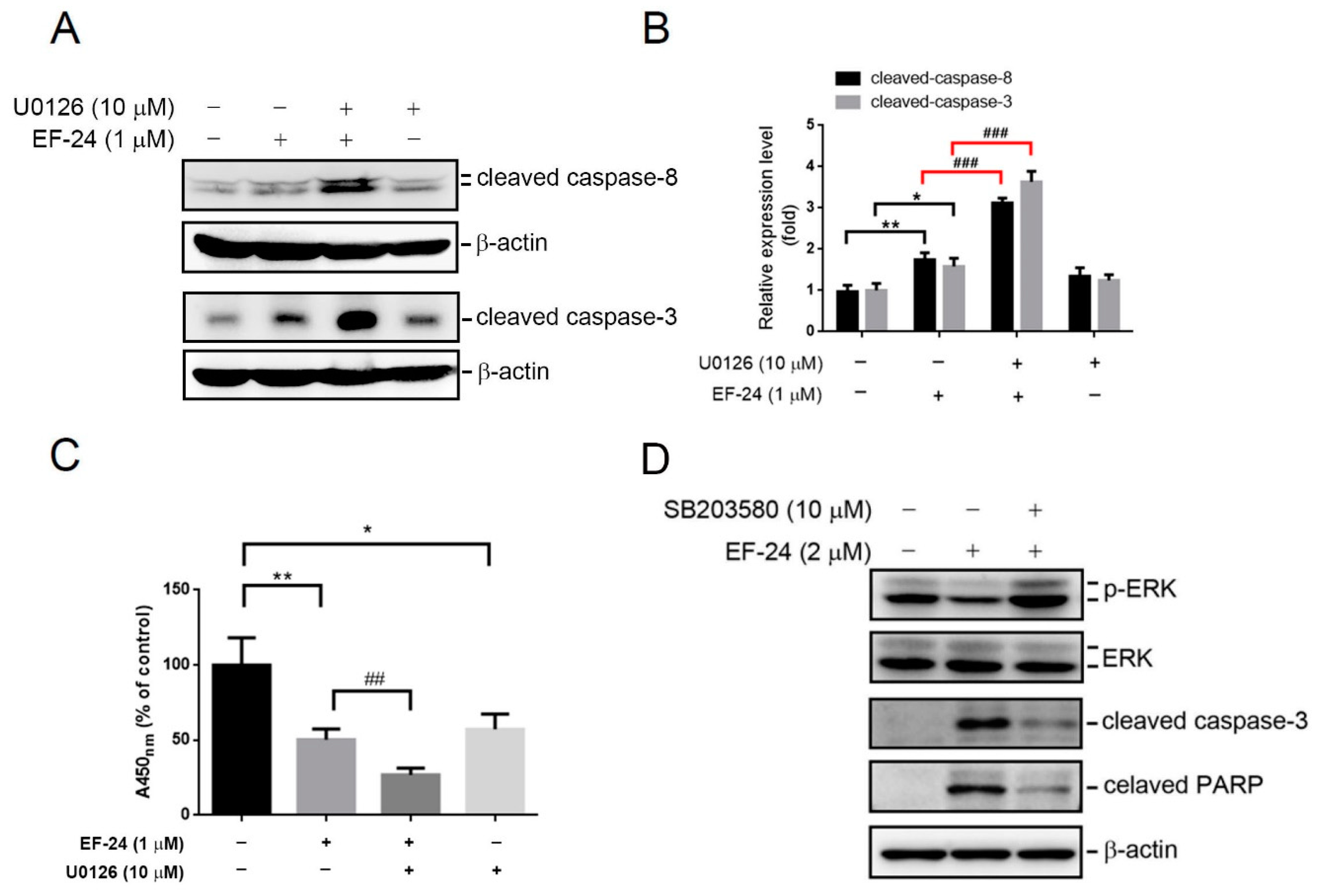
2.4. Relationship between p38 and ERK Activation in EF-24-Triggered Apoptosis of HL-60 Cells
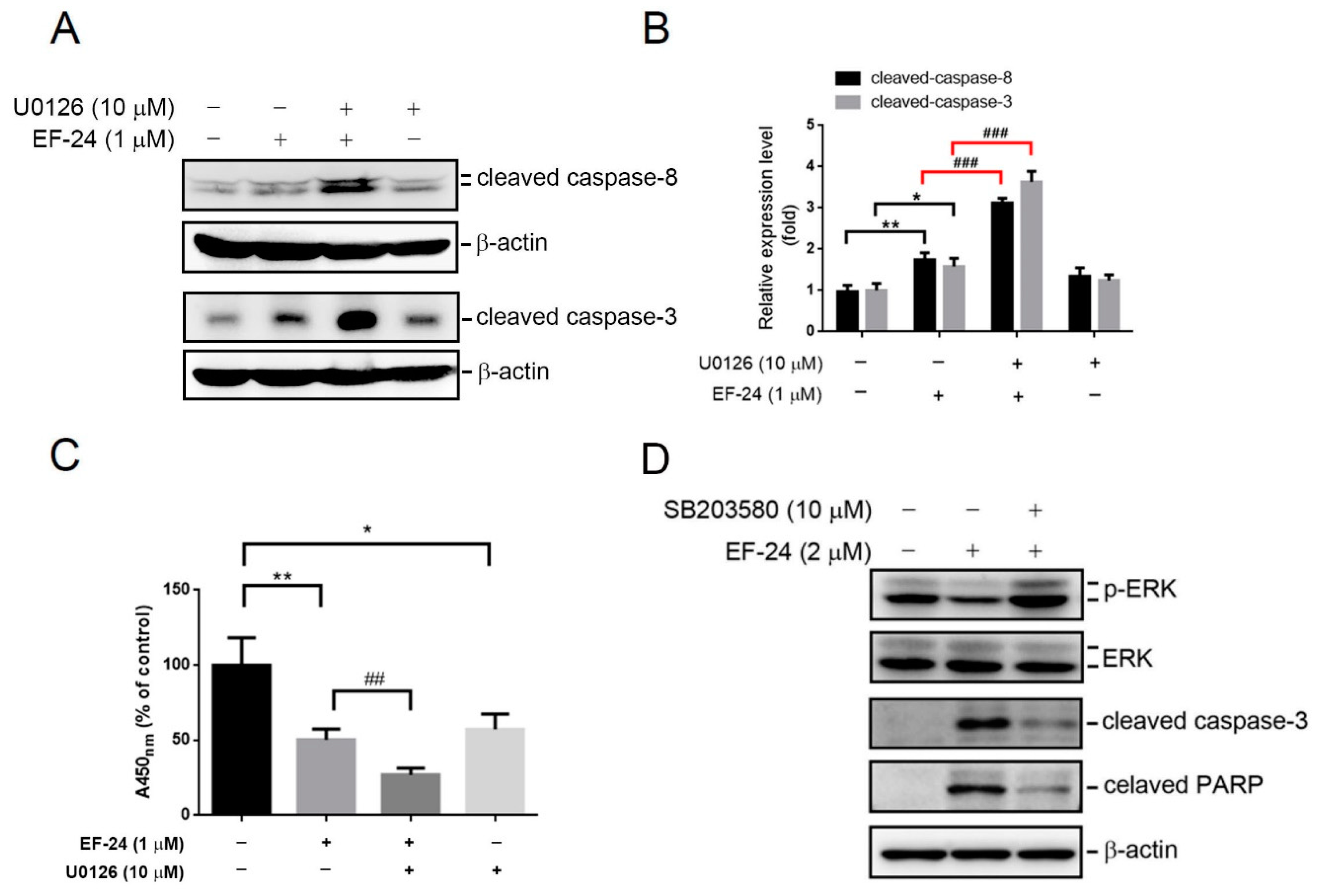
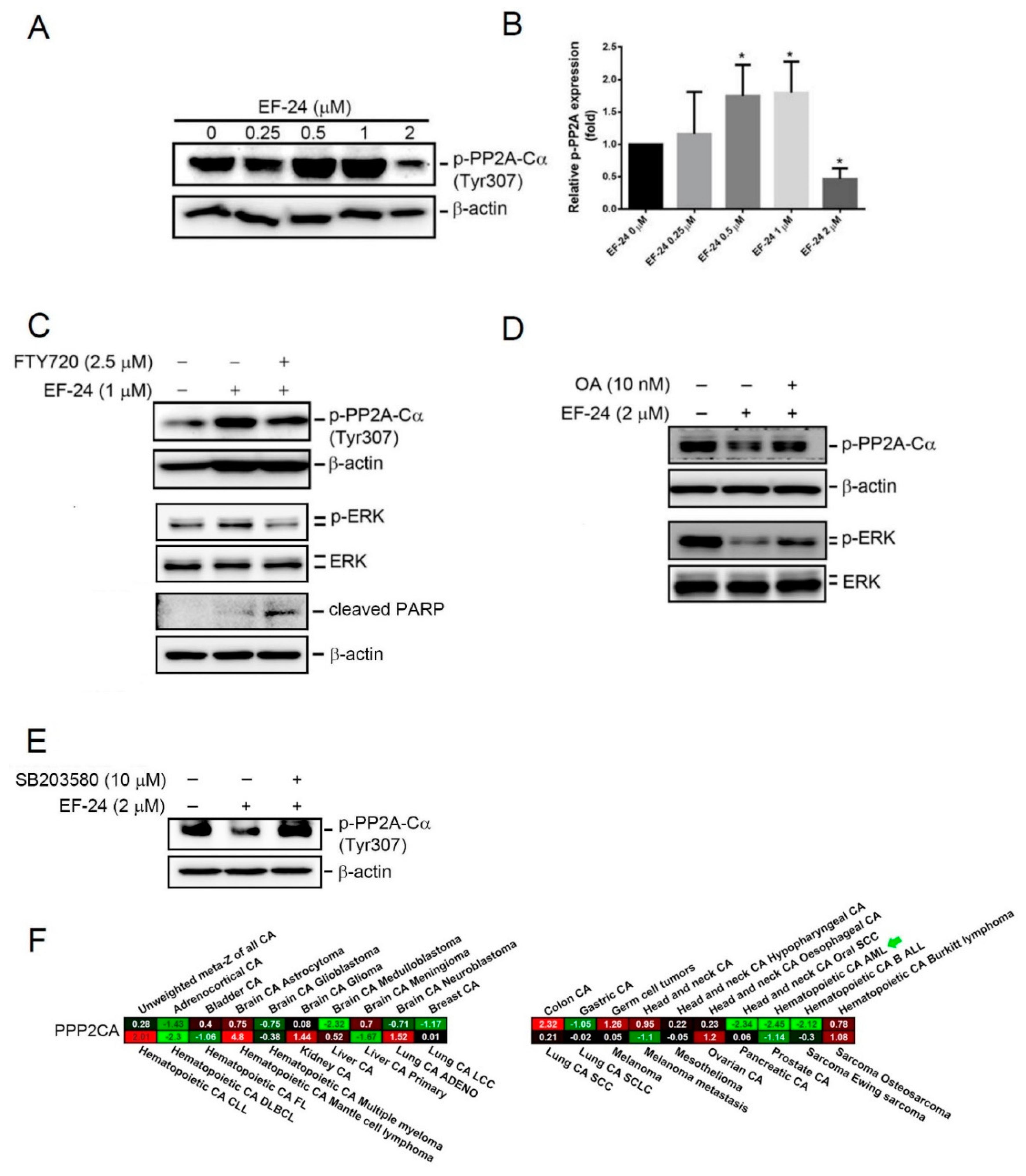
2.5. EF-24-Induced p38 Activation Negatively Regulates ERK Activity in a PP2A-Dependent Manner
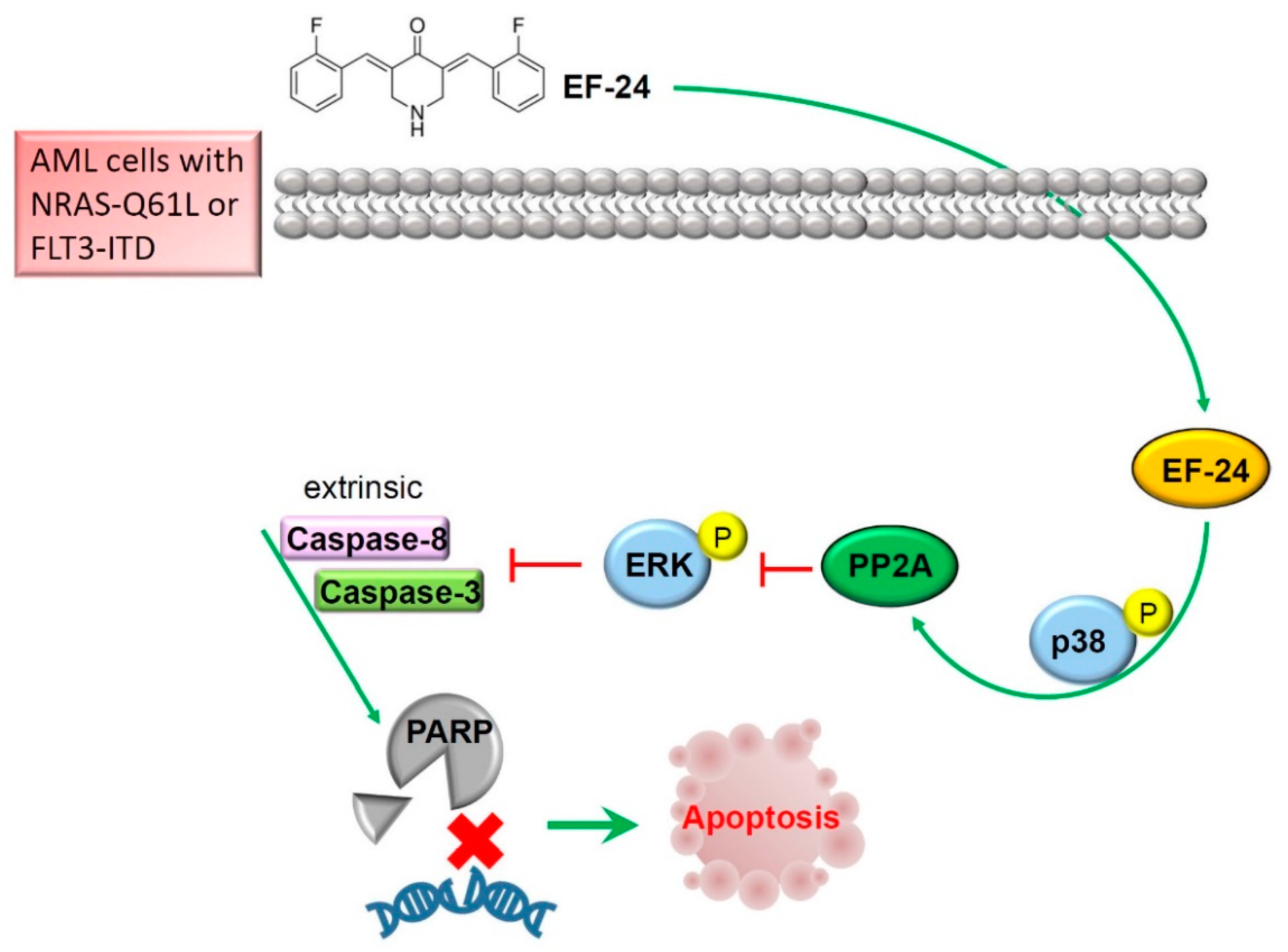
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Chemicals and Reagents
4.3. Cytotoxicity Assay
4.4. Fluorescence-Activated Cell Sorting Analysis of the Cell Cycle Distribution
4.5. Apoptosis Assays
4.6. Protein Lysate Preparation and Western Blot Analysis
4.7. Bioinformatics Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siveen, K.S.; Uddin, S.; Mohammad, R.M. Targeting acute myeloid leukemia stem cell signaling by natural products. Mol. Cancer 2017, 16, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015, 35, 645–651. [Google Scholar] [PubMed]
- Wang, M.; Jiang, S.; Zhou, L.; Yu, F.; Ding, H.; Li, P.; Zhou, M.; Wang, K. Potential Mechanisms of Action of Curcumin for Cancer Prevention: Focus on Cellular Signaling Pathways and miRNAs. Int. J. Biol. Sci. 2019, 15, 1200–1214. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. Aaps. J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer Activity of Curcumin and Its Analogues: Preclinical and Clinical Studies. Cancer Investig. 2017, 35, 1–22. [Google Scholar] [CrossRef]
- Heger, M. Drug screening: Don’t discount all curcumin trial data. Nature 2017, 543, 40. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of EF24, a Novel Curcumin Analog: A Review. Front. Oncol. 2018, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Hatamipour, M.; Ramezani, M.; Tabassi, S.A.S.; Johnston, T.P.; Ramezani, M.; Sahebkar, A. Demethoxycurcumin: A naturally occurring curcumin analogue with antitumor properties. J. Cell Physiol. 2018, 233, 9247–9260. [Google Scholar] [CrossRef]
- Reid, J.M.; Buhrow, S.A.; Gilbert, J.A.; Jia, L.; Shoji, M.; Snyder, J.P.; Ames, M.M. Mouse pharmacokinetics and metabolism of the curcumin analog, 4-piperidinone,3,5-bis[(2-fluorophenyl)methylene]-acetate(3E,5E) (EF-24; NSC 716993). Cancer Chemother. Pharmacol. 2014, 73, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Du, Y.; Thomas, S.L.; Zhao, J.; Sun, S.Y.; Khuri, F.R.; Wang, C.Y.; Shoji, M.; Sun, A.; Snyder, J.P.; et al. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol. Pharmacol. 2008, 74, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.; Zhong, D.; Zhou, W.; Malik, S.; Liotta, D.; Snyder, J.P.; Hamel, E.; Giannakakou, P. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle 2008, 7, 2409–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.P.; Wang, S.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple anticancer activities of EF24, a novel curcumin analog, on human ovarian carcinoma cells. Reprod Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.; Zhao, J.; Li, Z.; Lou, B.; Du, Y.; Purcell, J.; Snyder, J.P.; Khuri, F.R.; Liotta, D.; Fu, H. Activation of the p38 pathway by a novel monoketone curcumin analog, EF24, suggests a potential combination strategy. Biochem. Pharmacol. 2010, 80, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.J.; Williams, C.S. Protein Phosphatase 2A in the Regulation of Wnt Signaling, Stem Cells, and Cancer. Genes (Basel) 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.W.; Yan, L.; Yang, E. Phosphatases and regulation of cell death. Methods Enzymol. 2008, 446, 237–257. [Google Scholar]
- Martin, R.; Stonyte, V.; Lopez-Aviles, S. Protein Phosphatases in G1 Regulation. Int. J. Mol. Sci. 2020, 21, 395. [Google Scholar] [CrossRef] [Green Version]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef]
- Remmerie, M.; Janssens, V. PP2A: A Promising Biomarker and Therapeutic Target in Endometrial Cancer. Front. Oncol. 2019, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Dun, M.D.; Lee, E.M.; Harrison, C.; Kahl, R.; Flanagan, H.; Panicker, N.; Mashkani, B.; Don, A.S.; Morris, J.; et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget 2016, 7, 47465–47478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Shen, G.; Khor, T.O.; Kim, J.H.; Kong, A.N. Curcumin inhibits Akt/mammalian target of rapamycin signaling through protein phosphatase-dependent mechanism. Mol. Cancer Ther. 2008, 7, 2609–2620. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Xu, B.; Beevers, C.S.; Odaka, Y.; Chen, L.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; et al. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis 2012, 33, 868–875. [Google Scholar] [CrossRef]
- Chien, M.H.; Yang, W.E.; Yang, Y.C.; Ku, C.C.; Lee, W.J.; Tsai, M.Y.; Lin, C.W.; Yang, S.F. Dual Targeting of the p38 MAPK-HO-1 Axis and cIAP1/XIAP by Demethoxycurcumin Triggers Caspase-Mediated Apoptotic Cell Death in Oral Squamous Cell Carcinoma Cells. Cancers (Basel) 2020, 12, 703. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhong, J.; Yan, L.; Li, J.; Wang, H.; Wen, Y.; Zhao, Y. Curcumin exerts antitumor effects in retinoblastoma cells by regulating the JNK and p38 MAPK pathways. Int. J. Mol. Med. 2016, 38, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.H.; Dai, H.P.; Shen, Q.; Ji, O.; Zhang, Q.; Zhai, Y.L. Curcumin induces apoptosis and suppresses invasion through MAPK and MMP signaling in human monocytic leukemia SHI-1 cells. Pharm. Biol. 2016, 54, 1303–1311. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Lunghi, P.; Tabilio, A.; Dall’Aglio, P.P.; Ridolo, E.; Carlo-Stella, C.; Pelicci, P.G.; Bonati, A. Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia 2003, 17, 1783–1793. [Google Scholar] [CrossRef]
- Silverstein, A.M.; Barrow, C.A.; Davis, A.J.; Mumby, M.C. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. USA 2002, 99, 4221–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Martin, B.L.; Brautigan, D.L. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992, 257, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug therapy for acute myeloid leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh, E.; Shotorbani, S.S.; Baradaran, B. Herbal medicine as inducers of apoptosis in cancer treatment. Adv. Pharm. Bull. 2014, 4, 421–427. [Google Scholar]
- Wang, Z.Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef] [Green Version]
- Zou, P.; Xia, Y.; Chen, W.; Chen, X.; Ying, S.; Feng, Z.; Chen, T.; Ye, Q.; Wang, Z.; Qiu, C.; et al. EF24 induces ROS-mediated apoptosis via targeting thioredoxin reductase 1 in gastric cancer cells. Oncotarget 2016, 7, 18050–18064. [Google Scholar] [CrossRef] [Green Version]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Rincon, R.; Cristobal, I.; Zazo, S.; Arpi, O.; Menendez, S.; Manso, R.; Lluch, A.; Eroles, P.; Rovira, A.; Albanell, J.; et al. PP2A inhibition determines poor outcome and doxorubicin resistance in early breast cancer and its activation shows promising therapeutic effects. Oncotarget 2015, 6, 4299–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristobal, I.; Manso, R.; Rincon, R.; Carames, C.; Senin, C.; Borrero, A.; Martinez-Useros, J.; Rodriguez, M.; Zazo, S.; Aguilera, O.; et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol. Cancer Ther. 2014, 13, 938–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood 2014, 124, 3947–3955. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Tu, C.; Ma, Y.; Ye, P.; Shao, X.; Yang, Z.; Fang, Y. Curcumin analog EF24 induces apoptosis and downregulates the mitogen activated protein kinase/extracellular signal-regulated signaling pathway in oral squamous cell carcinoma. Mol. Med. Rep. 2017, 16, 4927–4933. [Google Scholar] [CrossRef]
- Hwang, D.; Kim, M.; Park, H.; Jeong, M.I.; Jung, W.; Kim, B. Natural Products and Acute Myeloid Leukemia: A Review Highlighting Mechanisms of Action. Nutrients 2019, 11, 1010. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Liu, Q.; Hofmann, P.A. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2204–H2212. [Google Scholar] [CrossRef] [Green Version]
- Westermarck, J.; Li, S.P.; Kallunki, T.; Han, J.; Kahari, V.M. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol. Cell. Biol. 2001, 21, 2373–2383. [Google Scholar] [CrossRef] [Green Version]
- Avdi, N.J.; Malcolm, K.C.; Nick, J.A.; Worthen, G.S. A role for protein phosphatase-2A in p38 mitogen-activated protein kinase-mediated regulation of the c-Jun NH(2)-terminal kinase pathway in human neutrophils. J. Biol. Chem. 2002, 277, 40687–40696. [Google Scholar] [CrossRef] [Green Version]
- Chien, M.H.; Chow, J.M.; Lee, W.J.; Chen, H.Y.; Tan, P.; Wen, Y.C.; Lin, Y.W.; Hsiao, P.C.; Yang, S.F. Tricetin Induces Apoptosis of Human Leukemic HL-60 Cells through a Reactive Oxygen Species-Mediated c-Jun N-Terminal Kinase Activation Pathway. Int. J. Mol. Sci. 2017, 18, 1667. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Cho, T.J.; Woo, B.H.; Choi, K.U.; Lee, C.H.; Ryu, M.H.; Park, H.R. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Arch. Oral Biol. 2012, 57, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gao, S.; Yang, Y.; Zhao, X.; Fan, Y.; Ma, W.; Yang, D.; Yang, A.; Yu, Y. Antitumor activity of curcumin by modulation of apoptosis and autophagy in human lung cancer A549 cells through inhibiting PI3K/Akt/mTOR pathway. Oncol. Rep. 2018, 39, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.L.; Li, J.; Qin, Z.H.; Liang, Z.Q. Autophagic and apoptotic mechanisms of curcumin-induced death in K562 cells. J. Asian Nat. Prod. Res. 2009, 11, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, T.; Sun, H.; Huang, B. Study on the Relationship between Autophagy and Apoptosis in A549 Cells Induced by Curcumin Analogue EF24. Chin. J. Cell Biol. 2012, 6, 590–596. [Google Scholar]
- Li, N.; Wen, S.; Chen, G.; Wang, S. Antiproliferative potential of piperine and curcumin in drug-resistant human leukemia cancer cells are mediated via autophagy and apoptosis induction, S-phase cell cycle arrest and inhibition of cell invasion and migration. J. BUON 2020, 25, 401–406. [Google Scholar]
- Chien, M.H.; Lee, W.J.; Yang, Y.C.; Tan, P.; Pan, K.F.; Liu, Y.C.; Tsai, H.C.; Hsu, C.H.; Wen, Y.C.; Hsiao, M.; et al. N-alpha-acetyltransferase 10 protein promotes metastasis by stabilizing matrix metalloproteinase-2 protein in human osteosarcomas. Cancer Lett. 2018, 433, 86–98. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, P.-C.; Chang, J.-H.; Lee, W.-J.; Ku, C.-C.; Tsai, M.-Y.; Yang, S.-F.; Chien, M.-H. The Curcumin Analogue, EF-24, Triggers p38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells. Cancers 2020, 12, 2163. https://doi.org/10.3390/cancers12082163
Hsiao P-C, Chang J-H, Lee W-J, Ku C-C, Tsai M-Y, Yang S-F, Chien M-H. The Curcumin Analogue, EF-24, Triggers p38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells. Cancers. 2020; 12(8):2163. https://doi.org/10.3390/cancers12082163
Chicago/Turabian StyleHsiao, Pei-Ching, Jer-Hwa Chang, Wei-Jiunn Lee, Chia-Chi Ku, Meng-Ying Tsai, Shun-Fa Yang, and Ming-Hsien Chien. 2020. "The Curcumin Analogue, EF-24, Triggers p38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells" Cancers 12, no. 8: 2163. https://doi.org/10.3390/cancers12082163
APA StyleHsiao, P.-C., Chang, J.-H., Lee, W.-J., Ku, C.-C., Tsai, M.-Y., Yang, S.-F., & Chien, M.-H. (2020). The Curcumin Analogue, EF-24, Triggers p38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells. Cancers, 12(8), 2163. https://doi.org/10.3390/cancers12082163