Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery

 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Choice of Cells for Targeted Drug Delivery
2.1. Tumor-Infiltrating Lymphocytes
2.2. Engineered T Cells
2.3. NK Cells
2.4. B Cell-Based Cancer Immunotherapy
2.5. iPSC-Based Cancer Immunotherapy
2.6. Macrophage-Based Cancer Immunotherapy
3. Vector Design and Gene Transfer for Engineered Cells
3.1. Multicistronic Vector Design
3.2. Multiple Promoter Systems and Co-Transduction
3.3. Non-Viral Gene Delivery Methods
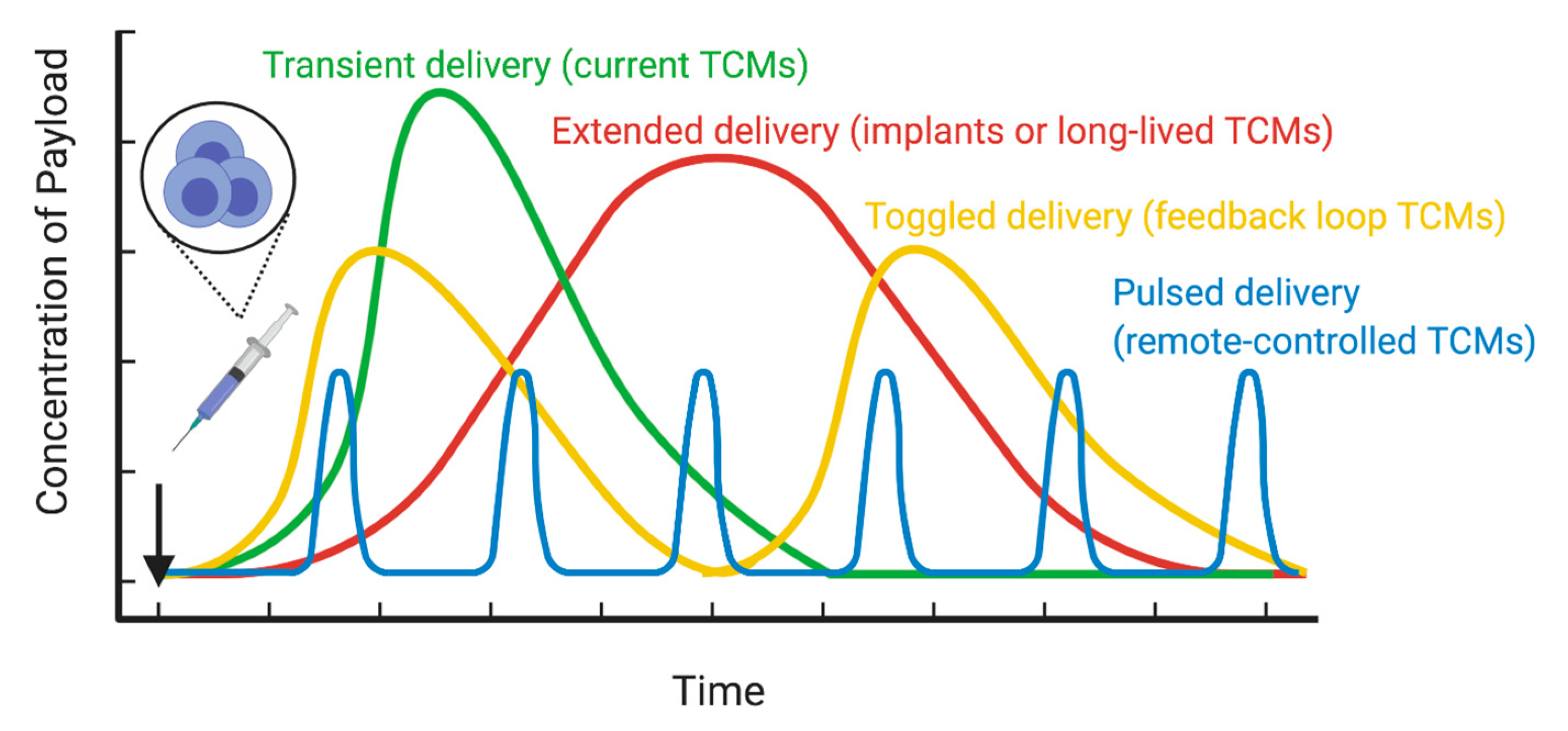
4. Approaches to Engineering Cells Inside the Patient
4.1. Transposase Delivery
4.2. mRNA Delivery
4.3. Cellular Implants
5. Cellular Delivery of Therapeutic Antibodies and Their Derivatives
5.1. CAR T Cells Secreting Antibodies to PDL-1/PD-1
5.2. CAR T Cells Secreting Antibodies to CTLA4
5.3. Cells that Disrupt the CD47–SIRPα Signaling Axis
5.4. Cellular Delivery of Antibodies Against Tumor-Associated Antigens
6. Delivery of Cellular-Modulating Agents in Cancer
6.1. TCMs Expressing Tumor Suppressor Proteins
6.2. Cytokines in the TME
6.3. Cytokines that Promote T Cell Persistence
6.4. TCMs that Prime Immune Effectors
7. Enzyme Delivery Strategies
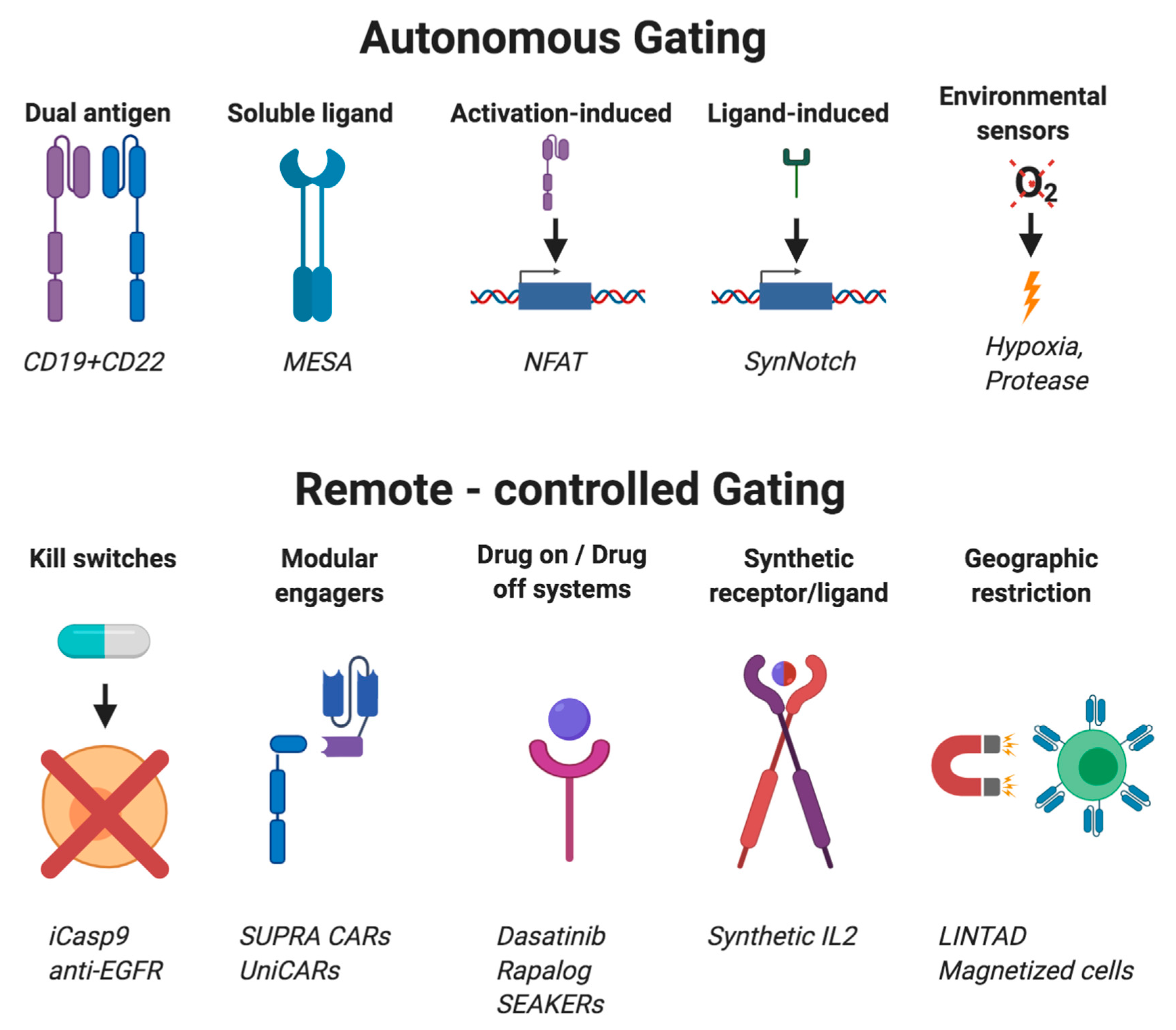
8. Cellular Gating Strategies
8.1. Autonomous Gating Systems
8.2. Activation-Dependent Systems
8.3. Activation-Independent Systems
8.4. Remote-Controlled Gating Systems
8.5. Challenges of Gated Systems in TCMs
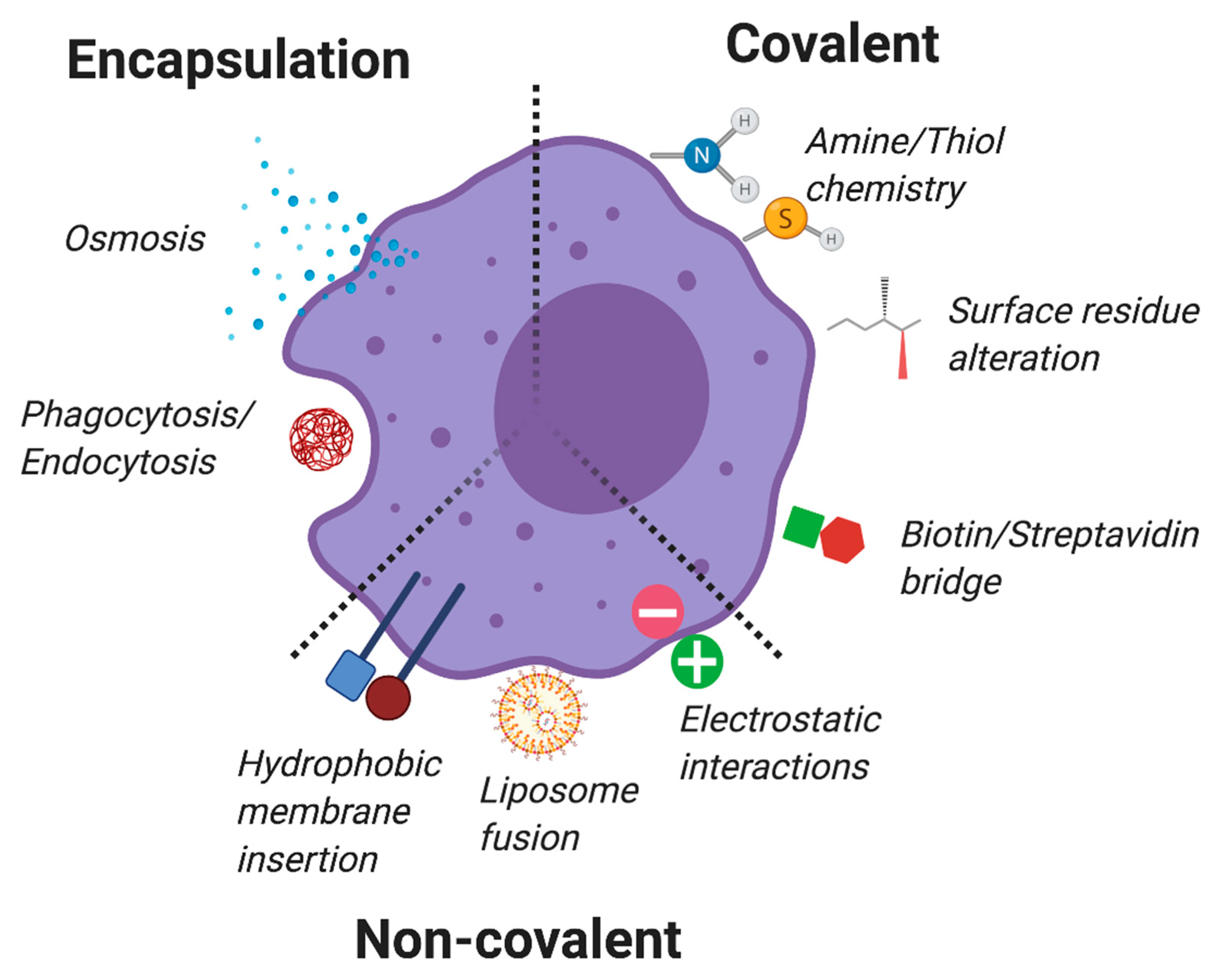
9. Non-Genetic Engineered TCs
9.1. Intracellular Encapsulation
9.2. Non-Covalent Surface Modifications
9.3. Covalent Membrane Conjugations
10. Additional Considerations and Applications in the Clinical Use of TCMs
10.1. Mesenchymal Stem Cell (MSC)-Based Strategies for Cancer Treatment
10.2. TCM for Autoimmune Disease
10.3. TCM for Neurological Disorders
10.4. Other Disease Targets of TCM
11. Conclusions/Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Ferguson, F.M.; Gray, N. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2017, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.H.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Claesen, J.; Fischbach, M.A. Synthetic Microbes as Drug Delivery Systems. ACS Synth. Boil. 2014, 4, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Lockney, D.; Franzen, S.; Lommel, S. Viruses as Nanomaterials for Drug Delivery. Methods Mol. Biol. 2011, 726, 207–221. [Google Scholar]
- Quintarelli, C.; Vera, J.F.; Savoldo, B.; Attianese, G.M.P.G.; Pulé, M.; Foster, A.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood 2007, 110, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, B.; Liu, K.; Dudley, M.E.; Johnson, L.A.; Kaiser, A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Matsuda, K.; Robbins, P.F.; et al. Adoptive Cell Therapy for Patients with Melanoma, Using Tumor-Infiltrating Lymphocytes Genetically Engineered to Secrete Interleukin-2. Hum. Gene Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, C.; Haymaker, C.L.; Hurwitz, M.E.; Kluger, H.; Tetzlaff, M.; Jackson, N.; Gergel, I.; Tagliaferri, M.A.; Zalevsky, J.; Hoch, U.; et al. Effect of a novel IL-2 cytokine immune agonist (NKTR-214) on proliferating CD8+ T cells and PD-1 expression on immune cells in the tumor microenvironment in patients with prior checkpoint therapy. J. Clin. Oncol. 2017, 35, 2545. [Google Scholar] [CrossRef]
- Butler, M.O.; Friedlander, P.; Milstein, M.I.; Mooney, M.M.; Metzler, G.; Murray, A.P.; Tanaka, M.; Berezovskaya, A.; Imataki, O.; Drury, L.; et al. Establishment of Antitumor Memory in Humans Using in Vitro-Educated CD8+ T Cells. Sci. Transl. Med. 2011, 3, 80ra34. [Google Scholar] [CrossRef] [Green Version]
- Carroll, R.G.; Carpenito, C.; Shan, X.; Danet-Desnoyers, G.; Liu, R.; Jiang, S.; Albelda, S.M.; Golovina, T.; Coukos, G.; Riley, J.L.; et al. Distinct Effects of IL-18 on the Engraftment and Function of Human Effector CD8+ T Cells and Regulatory T Cells. PLoS ONE 2008, 3, e3289. [Google Scholar] [CrossRef]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Hsu, C.; Hughes, M.S.; Zheng, Z.; Bray, R.B.; Rosenberg, S.A.; Morgan, R.A. Primary Human T Lymphocytes Engineered with a Codon-Optimized IL-15 Gene Resist Cytokine Withdrawal-Induced Apoptosis and Persist Long-Term in the Absence of Exogenous Cytokine. J. Immunol. 2005, 175, 7226–7234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Varela-Rohena, A.; Molloy, P.E.; Dunn, S.M.; Li, Y.; Suhoski, M.M.; Carroll, R.G.; Milicic, A.; Mahon, T.; Sutton, D.H.; Laugel, B.E.; et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat. Med. 2008, 14, 1390–1395. [Google Scholar] [CrossRef] [Green Version]
- Malviya, M.; Saoudi, A.; Bauer, J.; Fillatreau, S.; Liblau, R. Treatment of experimental autoimmune encephalomyelitis with engineered bi-specific Foxp3+ regulatory CD4+ T cells. J. Autoimmun. 2020, 108, 102401. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Barsan, V.; Mackall, C.L. Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert Opin. Boil. Ther. 2020, 20, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Moyes, K.W.; Lieberman, N.A.; Kreuser, S.A.; Chinn, H.; Winter, C.; Deutsch, G.; Hoglund, V.; Watson, R.; Crane, C.A. Genetically Engineered Macrophages: A Potential Platform for Cancer Immunotherapy. Hum. Gene Ther. 2017, 28, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shao, R.; Huang, H.; Wang, X.; Rong, Z.; Lin, Y. Engineering macrophages to phagocytose cancer cells by blocking the CD47/SIRPa axis. Cancer Med. 2019, 8, 4245–4253. [Google Scholar]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Lund, F.E.; Garvy, B.A.; Randall, T.D.; Harris, D.P. Regulatory Roles for Cytokine-Producing B Cells in Infection and Autoimmune Disease. Current Dir. Autoimmun. 2005, 8, 25–54. [Google Scholar] [CrossRef]
- Nguyen, K.-P.; Yang, W.; Saxena, R.; Thung, S.N.; Woo, S.L.; Chen, S.H. Role of NK and T cells in IL-12-induced anti-tumor response against hepatic colon carcinoma. Int. J. Cancer 1999, 81, 813–819. [Google Scholar] [CrossRef]
- Moutai, T.; Yamana, H.; Nojima, T.; Kitamura, D. A novel and effective cancer immunotherapy mouse model using antigen-specific B cells selected in vitro. PLoS ONE 2014, 9, e92732. [Google Scholar] [CrossRef] [Green Version]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4, eaax0644. [Google Scholar] [CrossRef]
- Johnson, M.J.; Laoharawee, K.; Lahr, W.S.; Webber, B.R.; Moriarity, B.S. Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci. Rep. 2018, 8, 12144. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.M.; Maarschalk, E.; O’Connell, R.M.; Wang, P.; Yang, L.; Baltimore, D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood 2009, 113, 1422–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Ma, W.; Lu, H.; Yuan, L.; An, L.; Wang, X.; Cheng, G.; Zuo, S. Modification of cytokine-induced killer cells with chimeric antigen receptors (CARs) enhances antitumor immunity to epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol. Immunother. 2015, 64, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Pizzitola, I.; Anjos-Afonso, F.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Vizcardo, R.; Klemen, N.D.; Islam, S.R.; Gurusamy, D.; Tamaoki, N.; Yamada, D.; Koseki, H.; Kidder, B.L.; Yu, Z.; Jia, L.; et al. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Rep. 2018, 22, 3175–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “Off-the-Shelf” Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, S. In Vitro Generation of Antigen-Specific T Cells from Induced Pluripotent Stem Cells of Antigen-Specific T Cell Origin. Methods Mol. Biol. 2016, 1393, 67–73. [Google Scholar] [CrossRef]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858.e4. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Xiong, Y.; Wu, D.; Nlle, V.; Schmitz, S.; Haso, W.; Kaiser, A.; Dropulic, B.; Orentas, R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Heyman, B.; Yang, Y. Chimeric Antigen Receptor Cell Therapy: Overcoming Obstacles to Battle Cancer. Cancers 2020, 12, 842. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; O’Rourke, D.M. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Pennisi, M.; Jain, T.; Santomasso, B.D.; Mead, E.; Wudhikarn, K.; Silverberg, M.L.; Batlevi, Y.; Shouval, R.; Devlin, S.M.; Batlevi, C.L.; et al. Comparing CAR T-cell toxicity grading systems: Application of the ASTCT grading system and implications for management. Blood Adv. 2020, 4, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Epperly, R.; Gottschalk, S.; Velasquez, M.P. A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front. Oncol. 2020, 10, 262. [Google Scholar] [CrossRef] [Green Version]
- Scarfò, I.; Maus, M.V. Unraveling the Signaling Balance of Activation and Exhaustion of CAR T Cells. Cancer Cell 2020, 37, 143–144. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.T.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.S.; Purdy, A.K. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011, 132, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Yu, J.; Caligiuri, M.A. Human natural killer cell development in secondary lymphoid tissues. Semin. Immunol. 2014, 26, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, L.; De Andrade, L.F.; Guillerey, C.; Lee, J.S.; Liu, J.; Souza-Fonseca-Guimaraes, F.; Hutchinson, D.S.; Kolesnik, T.B.; Nicholson, S.E.; Huntington, N.D.; et al. DNAM-1 Expression Marks an Alternative Program of NK Cell Maturation. Cell Rep. 2015, 11, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazetic, S.; Chang, C.; Houchins, J.P.; Lanier, L.L.; Phillips, J.H. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J. Immunol. 1996, 157, 4741–4745. [Google Scholar] [PubMed]
- Fauriat, C.; Long, E.O.; Ljunggren, H.-G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.-J.; Kaufman, D. Clinical-Scale Derivation of Natural Killer Cells from Human Pluripotent Stem Cells for Cancer Therapy. STEM CELLS Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Kaufman, D.S. Utilizing Chimeric Antigen Receptors to Direct Natural Killer Cell Activity. Front. Immunol. 2015, 6, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Maurice, Y.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Wang, G.; Huang, D.-S.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Boieri, M.; Ulvmoen, A.; Sudworth, A.; Lendrem, C.; Collin, M.; Dickinson, A.M.; Kveberg, L.; Inngjerdingen, M. IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. OncoImmunology 2017, 6, e1274478. [Google Scholar] [CrossRef] [Green Version]
- French, A.; Yang, C.-T.; Taylor, S.; Watt, S.; Carpenter, L. Human Induced Pluripotent Stem Cell-Derived B Lymphocytes Express sIgM and Can Be Generated via a Hemogenic Endothelium Intermediate. Stem Cells Dev. 2015, 24, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Sachamitr, P.; Leishman, A.J.; Davies, T.J.; Fairchild, P.J. Directed Differentiation of Human Induced Pluripotent Stem Cells into Dendritic Cells Displaying Tolerogenic Properties and Resembling the CD141+ Subset. Front. Immunol. 2018, 8, 1935. [Google Scholar] [CrossRef]
- Hong, D.; Ding, J.; Li, O.; He, Q.; Ke, M.; Zhu, M.; Liu, L.; Ou, W.-B.; He, Y.; Wu, Y. Human-induced pluripotent stem cell-derived macrophages and their immunological function in response to tuberculosis infection. Stem Cell Res. Ther. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.-I.; Koseki, H.; Kawamoto, H. Regeneration of Human Tumor Antigen-Specific T Cells from iPSCs Derived from Mature CD8 + T Cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Keller, B.C.; Makalou, N.; Sutton, R.E. Systematic Determination of the Packaging Limit of Lentiviral Vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Vile, R.G.; Russell, S.J. Retroviruses as vectors. Br. Med Bull. 1995, 51, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.M.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Huang, Q.; Fang, Y.; Wu, Y.F.A.J. FurinDB: A Database of 20-Residue Furin Cleavage Site Motifs, Substrates and Their Associated Drugs. Int. J. Mol. Sci. 2011, 12, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Milsom, M.D.; Shaffer, J.; Neuenfeldt, J.; Shaaban, A.F.; Margison, G.P.; Fairbairn, L.J.; Chinnasamy, N. Multicistronic lentiviral vectors containing the FMDV 2A cleavage factor demonstrate robust expression of encoded genes at limiting MOI. Virol. J. 2006, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Gurtu, V.; Yan, G.; Zhang, G. IRES Bicistronic Expression Vectors for Efficient Creation of Stable Mammalian Cell Lines. Biochem. Biophys. Res. Commun. 1996, 229, 295–298. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Xu, Z.; Ishii-Watabe, A.; Uchida, E.; Hayakawa, T. IRES-Dependent Second Gene Expression Is Significantly Lower Than Cap-Dependent First Gene Expression in a Bicistronic Vector. Mol. Ther. 2000, 1, 376–382. [Google Scholar] [CrossRef]
- De Felipe, P. Polycistronic Viral Vectors. Curr. Gene Ther. 2002, 2, 355–378. [Google Scholar] [CrossRef] [Green Version]
- Frimpong, K.; Spector, S.A. Cotransduction of nondividing cells using lentiviral vectors. Gene Ther. 2000, 7, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Wotherspoon, S.; Dolnikov, A.; Symonds, G.; Nordon, R.E. Susceptibility of Cell Populations to Transduction by Retroviral Vectors. J. Virol. 2004, 78, 5097–5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Loo, J.C.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2015, 25, R42–R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffett, H.F.; Coon, M.E.; Radtke, S.; Stephan, S.B.; McKnight, L.; Lambert, A.; Stoddard, B.L.; Kiem, H.P.; Stephan, M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, G.; Zhang, L.; Zhao, Q. Building Potent Chimeric Antigen Receptor T Cells with CRISPR Genome Editing. Front. Immunol. 2019, 10, 456. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2016, 27, 154–157. [Google Scholar] [CrossRef]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Coon, M.E.; Stephan, S.B.; Gupta, V.; Kealey, C.P.; Stephan, M.T. Nitinol thin films functionalized with CAR-T cells for the treatment of solid tumours. Nat. Biomed. Eng. 2019, 4, 195–206. [Google Scholar] [CrossRef]
- Stephan, S.B.; Taber, A.M.; Jileaeva, I.; Pegues, E.P.; Sentman, C.L.; Stephan, M.T. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 2014, 33, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Investig. 2017, 127, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Dane, A.P.; Swanson, A.; Alexander, I.E.; Ginn, S.L. Bidirectional promoter interference between two widely used internal heterologous promoters in a late-generation lentiviral construct. Gene Ther. 2008, 15, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; Chai, Y.C.; VandenDriessche, T.; Chuah, M.K. Preclinical and clinical advances in transposon-based gene therapy. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The Emerging Role of In Vitro-Transcribed mRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Castiello, L.; Aricò, E.; D’Agostino, G.; Santodonato, L.; Belardelli, F. In situ Vaccination by Direct Dendritic Cell Inoculation: The Coming of Age of an Old Idea? Front. Immunol. 2019, 10, 2303. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Stone, R.M.; Uhl, L.; Neuberg, N.; Joyce, R.; Levine, J.D.; Arnason, J.; McMasters, M.; Luptakova, K.; Jain, S.; et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci. Transl. Med. 2016, 8, 368ra171. [Google Scholar] [CrossRef] [Green Version]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Firer, M.A.; Gellerman, G. Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol. 2012, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Cham, J.; Zhang, L.; Fong, G.; Kwek, S.S.; Klinger, M.; Faham, M.; Fong, L. Immune Toxicities Elicted by CTLA-4 Blockade in Cancer Patients Are Associated with Early Diversification of the T-cell Repertoire. Cancer Res. 2016, 77, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Nikpoor, A.R.; Tavakkol-Afshari, J.; Sadri, K.; Jalali, S.A.; Jaafari, M.R. Improved tumor accumulation and therapeutic efficacy of CTLA-4-blocking antibody using liposome-encapsulated antibody: In vitro and in vivo studies. Nanomed. Nanotechnol. Boil. Med. 2017, 13, 2671–2682. [Google Scholar] [CrossRef]
- Zimmer, L.; Goldinger, S.; Hofmann, L.; Loquai, C.; Ugurel, S.; Thomas, I.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.; Göppner, D.; et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 210–225. [Google Scholar] [CrossRef]
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376. [Google Scholar] [CrossRef]
- Cherkassky, L.; Sadelain, M.; Prasad, S.; Invest, J.C.; Morello, A.; Villena-vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition Find the latest version: Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. 2016, 126, 3130–3144. [Google Scholar]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Zolov, S.N.; Rietberg, S.P.; Bonifant, C.L. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR–T cells. Cytotherapy 2018, 20, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Suarez, E.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor–Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Meng, T.; Zhao, Z.; Han, J.; Zhang, W.; Gao, F.; Cai, J. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 2017, 636, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Condomines, M.; Arnason, J.; Benjamin, R.; Gunset, G.; Plotkin, J.; Sadelain, M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS ONE 2015, 10, e0130518. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. UAS 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. mABs 2015, 7, 303–310. [Google Scholar] [CrossRef]
- Chao, W.M. The CD47-SIRP Pathway in Cancer Immune Evasion and potential Therapeutic Implications. Cell 2011, 24, 225–232. [Google Scholar]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.-P.; Levin, M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anti-cancer antibodies. Science 2014, 341, 1–13. [Google Scholar]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.-X.; Xu, M.M. CD47 blockade triggers T cell–mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Mathias, M.D.; Sockolosky, J.T.; Chang, A.Y.; Tan, K.S.; Liu, C.; Garcia, K.C.; Scheinberg, D.A. CD47 blockade enhances therapeutic activity of TCR mimic antibodies to ultra-low density cancer epitopes. Leukemia 2017, 31, 2254–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.J.; Dougan, M.; Ingram, J.R.; Pishesha, N.; Fang, T.; Momin, N.; Ploegh, H.L. Improved Antitumor Efficacy of Chimeric Antigen Receptor T Cells that Secrete Single-Domain Antibody Fragments. Cancer Immunol. Res. 2020, 8, 518–529. [Google Scholar] [CrossRef]
- Dacek, M.G.; Gardner, T.; Scheinberg, D. Potentiating Innate and Adaptive Immunity with Engineered CAR T Cells. In Proceedings of the Keystone Symposium on Cancer Therapy: Mechanistic Insight to Improve Clinical Benefit, Whistler, BC, Canada, 12 March 2019. [Google Scholar]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.A. Cardiac Toxicity of ErbB2-Targeted Therapies: What Do We Know? Clin. Breast Cancer 2008, 8, S114–S120. [Google Scholar] [CrossRef]
- Kakarla, S.; Gottschalk, S. CAR T Cells for Solid Tumors. Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Sun, W. Functional Mechanisms for Human Tumor Suppressors. J. Cancer 2010, 1, 136. [Google Scholar] [CrossRef] [PubMed]
- Boice, M.; Salloum, D.; Mourcin, F.; Sanghvi, V.; Amin, R.; Oricchio, E.; Jiang, M.; Mottok, A.; Denis-Lagache, N.; Ciriello, G.; et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 2016, 167, 405–418.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Textor, A.; Listopad, J.J.; Wuhrmann, L.L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2013, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wesa, A.K.; Galy, A. IL-1 beta induces dendritic cells to produce IL-12. Int. Immunol. 2001, 13, 1053–1061. [Google Scholar]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.L.; Hou, A.J.; Chen, Y.Y. Engineering primary T cells with chimeric antigen receptors for rewired responses to soluble ligands. Nat. Protoc. 2020, 15, 1507–1524. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Methods 2018, 14, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Tang, Q. Treg cells—The next frontier of cell therapy. Science 2018, 362, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Finkelstein, S.E.; Surman, D.R.; Lichtman, M.K.; Gattinoni, L.; Theoret, M.R.; Grewal, N.; Spiess, P.J.; Antony, P.A.; Palmer, D.C.; et al. IL-15 enhances thein vivoantitumor activity of tumor-reactive CD8+T Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1969–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2017, 32, 520–531. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Kren, N.P.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Pegram, H.J.; Park, J.H.; Brentjens, R.J. CD28z CARs and Armored CARs. Cancer J. 2014, 20, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Stephan, M.T.; Ponomarev, V.; Brentjens, R.J.; Chang, A.H.; Dobrenkov, K.V.; Heller, G.; Sadelain, M. T cell–encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med. 2007, 13, 1440–1449. [Google Scholar] [CrossRef]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; Van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing Antitumor Efficacy of Chimeric Antigen Receptor T Cells Through Constitutive CD40L Expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, N.F.; Purdon, T.J.; Van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e6. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Tahara, H.; Robbins, P.D.; Storkus, W.J.; Clarke, M.R.; Nalesnik, M.A.; Lotze, M.T. Cancer immunotherapy of established tumors with IL-Effective delivery by genetically engineered fibroblasts. J. Immunol. 1995, 155, 1393–1403. [Google Scholar]
- Kang, W.K.; Park, C.; Yoon, H.L.; Kim, W.S.; Yoon, S.-S.; Lee, M.H.; Park, K.; Kim, K.; Jeong, H.S.; Kim, J.-A.; et al. Interleukin 12 Gene Therapy of Cancer by Peritumoral Injection of Transduced Autologous Fibroblasts: Outcome of a Phase I Study. Hum. Gene Ther. 2001, 12, 671–684. [Google Scholar] [CrossRef]
- Tatsumi, T.; Huang, J.; Gooding, W.E.; Gambotto, A.; Robbins, P.D.; Vujanovic, N.L.; Alber, S.M.; Watkins, S.C.; Okada, H.; Storkus, W.J. Intratumoral delivery of dendritic cells engineered to secrete both interleukin (IL)-12 and IL-18 effectively treats local and distant disease in association with broadly reactive Tc1-type immunity. Cancer Res. 2003, 63, 6378–6386. [Google Scholar]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Koneru, M.; Purdon, T.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Ward, J.E.; McNeel, D.G. GVAX: An allogeneic, whole-cell, GM-CSF-secreting cellular immunotherapy for the treatment of prostate cancer. Expert Opin. Boil. Ther. 2007, 7, 1893–1902. [Google Scholar] [CrossRef]
- Garaude, J.; Kent, A.; Van Rooijen, N.; Blander, J.M. Simultaneous Targeting of Toll- and Nod-Like Receptors Induces Effective Tumor-Specific Immune Responses. Sci. Transl. Med. 2012, 4, 120ra16. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. J. Immunol. 2015, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Gardner, T.; Lee, P.J.; Wijewarnasuriya, D.; Kinarivala, D.; Bourne, C.; Kurtz, K.; Corless, B.; Dacek, M.; Mo, G.; Nguyen, K.; et al. A CAR T Cell that Creates Cytotoxic Small Molecules to Overcome Cancer Resistance Mechanisms. In Proceedings of the Keystone Symposium on Cancer Therapy: Mechanistic Insight to Improve Clinical Benefit, Whistler, BC, Canada, 12 March 2019. [Google Scholar]
- Bourne, C.; Gardner, T.; Romero-Pichardo, J.; Lee, P.; Dacek, M.; Tan, D.; Scheinberg, D. Mechanisms of Adoptive T Cell Micropharmacies. In Proceedings of the American Society of Gene and Cell Therapy Virtual Conference, 12–15 May 2020. Online, Abstract 31. [Google Scholar]
- Bagshawe, K.D. Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Expert Rev. Anticancer. Ther. 2006, 6, 1421–1431. [Google Scholar] [CrossRef]
- Basel, M.T.; Balivada, S.; Shrestha, T.B.; Seo, G.-M.; Pyle, M.M.; Tamura, M.; Bossmann, S.H.; Troyer, D.L. A Cell-Delivered and Cell-Activated SN38-Dextran Prodrug Increases Survival in a Murine Disseminated Pancreatic Cancer Model. Small 2012, 8, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Kan, O.; Day, D.; Iqball, S.; Burke, F.; Grimshaw, M.J.; Naylor, S.; Binley, K. Genetically modified macrophages expressing hypoxia regulated cytochrome P450 and P450 reductase for the treatment of cancer. Int. J. Mol. Med. 2011, 27, 173–180. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Latouche, J.-B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Uchibori, R.; Teruya, T.; Ido, H.; Ohmine, K.; Sehara, Y.; Urabe, M.; Mizukami, H.; Mineno, J.; Ozawa, K. Functional Analysis of an Inducible Promoter Driven by Activation Signals from a Chimeric Antigen Receptor. Mol. Ther. Oncolytics 2019, 12, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Daringer, N.M.; Dudek, R.M.; Schwarz, K.A.; Leonard, J.N. Modular Extracellular Sensor Architecture for Engineering Mammalian Cell-based Devices. ACS Synth. Boil. 2014, 3, 892–902. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Han, X.; Bryson, P.D.; Zhao, Y.; Cinay, G.E.; Li, S.; Guo, Y.; Siriwon, N.; Wang, P. Masked Chimeric Antigen Receptor for Tumor-Specific Activation. Mol. Ther. 2017, 25, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.C.; Pulé, M.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; Von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [Green Version]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Rojas, J.M.; Marcos, A.; Portilla, Y.; Stein, J.V.; Barber, D. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J. Nanobiotechnol. 2019, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.-J.; Wang, X.; Chien, S.; Wang, Y. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Saudemont, A.; Jespers, L.; Clay, T. Current Status of Gene Engineering Cell Therapeutics. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Csizmar, C.; Petersburg, J.R.; Wagner, C.R. Programming Cell-Cell Interactions through Non-genetic Membrane Engineering. Cell Chem. Boil. 2018, 25, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Andrade, B.; Seo, Y.; Kim, M.-J.; Zimmerman, S.C.; Kong, H. Engineering the Surface of Therapeutic “Living” Cells. Chem. Rev. 2018, 118, 1664–1690. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, H.; Peng, H.; Zhou, H.; Li, P.Y.; Langer, R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv. Drug Deliv. Rev. 2015, 91, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Chessa, L.; Leuzzi, V.; Plebani, A.; Soresina, A.; Micheli, R.; D’Agnano, D.; Venturi, T.; Molinaro, A.; Fazzi, E.; Marini, M.; et al. Intra-Erythrocyte Infusion of Dexamethasone Reduces Neurological Symptoms in Ataxia Teleangiectasia Patients: Results of a Phase 2 Trial. Orphanet J. Rare Dis. 2014, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunault-Berger, M.; Leguay, T.; Huguet, F.; Lepretre, S.; Deconinck, E.; Ojeda-Uribe, M.; Bonmati, C.; Escoffre-Barbe, M.; Bories, P.; Himberlin, C.; et al. A Phase 2 study of L-asparaginase encapsulated in erythrocytes in elderly patients with Philadelphia chromosome negative acute lymphoblastic leukemia: The GRASPALL/GRAALL-SA2-2008 study. Am. J. Hematol. 2015, 90, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Levene, M.; Coleman, D.G.; Kilpatrick, H.C.; Fairbanks, L.D.; Gangadharan, B.; Gasson, C.; Bax, B.E. Preclinical Toxicity Evaluation of Erythrocyte-Encapsulated Thymidine Phosphorylase in BALB/c Mice and Beagle Dogs: An Enzyme-Replacement Therapy for Mitochondrial Neurogastrointestinal Encephalomyopathy. Toxicol. Sci. 2012, 131, 311–324. [Google Scholar] [CrossRef]
- Choi, M.-R.; Stanton-Maxey, K.J.; Stanley, J.K.; Levin, C.S.; Bardhan, R.; Akin, D.; Badve, S.S.; Sturgis, J.; Robinson, J.; Bashir, R.; et al. A Cellular Trojan Horse for Delivery of Therapeutic Nanoparticles into Tumors. Nano Lett. 2007, 7, 3759–3765. [Google Scholar] [CrossRef]
- Dou, H.; Destache, C.J.; Morehead, J.R.; Mosley, R.L.; Boska, M.D.; Kingsley, J.; Gorantla, S.; Poluektova, L.Y.; Nelson, J.A.; Chaubal, M.; et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood 2006, 108, 2827–2835. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Chu, D.; Wang, Z. Leukocyte-mediated Delivery of Nanotherapeutics in Inflammatory and Tumor Sites. Theranostics 2017, 7, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Cole, C.; Qiao, J.; Kottke, T.; Diaz, R.M.; Ahmed, A.; Sanchez-Perez, L.; Brunn, G.; Thompson, J.; Chester, J.; Vile, R.G. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat. Med. 2005, 11, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Paulick, M.G.; Bertozzi, C.R. The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Pulsipher, A.; Luo, W.; Mak, H.; Yousaf, M.N. Engineering Cell Surfaces via Liposome Fusion. Bioconjugate Chem. 2011, 22, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, H.; Han, Y.; Wang, K.; Dong, H.; Li, Y.; Shi, D.; Li, Y. Remodeling of Cellular Surfaces via Fast Disulfide–Thiol Exchange to Regulate Cell Behaviors. ACS Appl. Mater. Interfaces 2019, 11, 47750–47761. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Abraham, W.D.; Zheng, Y.; López, S.C.B.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 2015, 7, 291ra94. [Google Scholar] [CrossRef] [Green Version]
- Lutterotti, A.; Yousef, S.; Sputtek, A.; Stürner, K.H.; Stellmann, J.-P.; Breiden, P.; Reinhardt, S.; Schulze, C.; Bester, M.; Heesen, C.; et al. Antigen-Specific Tolerance by Autologous Myelin Peptide-Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci. Transl. Med. 2013, 5, 188ra75. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.N. Biotinylation of Cell Surface Proteins. Bio. Protocol. 2012, 2. [Google Scholar] [CrossRef]
- Koyfman, A.Y.; Braun, G.B.; Reich, N.O. Cell-Targeted Self-Assembled DNA Nanostructures. J. Am. Chem. Soc. 2009, 131, 14237–14239. [Google Scholar] [CrossRef]
- Murciano, J.-C.; Medinilla, S.; Eslin, D.; Atochina, E.; Cines, D.B.; Muzykantov, V.R. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat. Biotechnol. 2003, 21, 891–896. [Google Scholar] [CrossRef]
- De Bank, P.; Kellam, B.; Kendall, D.A.; Shakesheff, K.M. Surface engineering of living myoblasts via selective periodate oxidation. Biotechnol. Bioeng. 2003, 81, 800–808. [Google Scholar] [CrossRef]
- Kayser, H.; Zeitler, R.; Kannicht, C.; Grunow, D.; Nuck, R.; Reutter, W. Biosynthesis of a nonphysiological sialic acid in different rat organs, using N-propanoyl-D-hexosamines as precursors. J. Boil. Chem. 1992, 267, 16934–16938. [Google Scholar]
- Wang, X.; Lang, S.; Tian, Y.; Zhang, J.; Yan, X.; Fang, Z.; Weng, J.; Lu, N.; Wu, X.; Li, T.; et al. Glycoengineering of Natural Killer Cells with CD22 Ligands for Enhanced Anticancer Immunotherapy. ACS Central Sci. 2020, 6, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.-Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K.; et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Wayteck, L.; Dewitte, H.; De Backer, L.; Breckpot, K.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. Hitchhiking nanoparticles: Reversible coupling of lipid-based nanoparticles to cytotoxic T lymphocytes. Biomaterials 2016, 77, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Siegler, E.L.; Kim, Y.J.; Chen, X.; Siriwon, N.; Mac, J.; Rohrs, J.A.; Bryson, P.D.; Wang, P. Combination Cancer Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells as Drug Carriers. Mol. Ther. 2017, 25, 2607–2619. [Google Scholar] [CrossRef] [Green Version]
- Siriwon, N.; Kim, Y.J.; Siegler, E.L.; Chen, X.; Liu, Y.; Wang, P.; Rohrs, J.A. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunol. Res. 2018, 6, 812–824. [Google Scholar] [CrossRef] [Green Version]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef] [Green Version]
- Amara, I.; Touati, W.; Beaune, P.; De Waziers, I. Mesenchymal stem cells as cellular vehicles for prodrug gene therapy against tumors. Biochimie 2014, 105, 4–11. [Google Scholar] [CrossRef]
- Marofi, F.; Vahedi, G.; Biglari, A.; Esmaeilzadeh, A.; Athari, S.S. Mesenchymal Stromal/Stem Cells: A New Era in the Cell-Based Targeted Gene Therapy of Cancer. Front. Immunol. 2017, 8, 1770. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Park, H.S.; Cho, M.L.; Oh, H.J.; Cho, Y.G.; Min, S.Y.; Chung, B.H.; Lee, J.W.; Kim, H.Y.; Cho, S.G. Transforming growth factor beta-transduced mesenchymal stem cells ameliorate experimental autoimmune arthritis through reciprocal regulation of Treg/Th17 cells and osteoclastogenesis. Arthritis Rheum. 2011, 63, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, C.; Deng, X.; Chen, L.; Hao, S.; Ma, L. Enhanced alleviation of aGVHD by TGF-beta1-modified mesenchymal stem cells in mice through shifting MPhi into M2 phenotype and promoting the differentiation of Treg cells. J. Cell Mol. Med. 2020, 24, 1684–1699. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cheng, Y.; Baylink, D.J.; Wasnik, S.; Goel, G.; Huang, M.; Cao, H.; Qin, X.; Lau, K.-H.W.; Chan, C.; et al. In Vivo Generation of Gut-Homing Regulatory T Cells for the Suppression of Colitis. J. Immunol. 2019, 202, 3447–3457. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, N.; Blesch, A.; Fouad, K. BDNF: The career of a multifaceted neurotrophin in spinal cord injury. Exp. Neurol. 2012, 238, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Haney, M.J.; Gupta, R.; Bohnsack, J.P.; He, Z.; Kabanov, A.V.; Batrakova, E.V. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS ONE 2014, 9, e106867. [Google Scholar] [CrossRef]
- Manning, E.; Pham, S.M.; Li, S.; Vazquez-Padron, R.I.; Mathew, J.M.; Ruiz, P.; Salgar, S.K. Interleukin-10 Delivery via Mesenchymal Stem Cells: A Novel Gene Therapy Approach to Prevent Lung Ischemia–Reperfusion Injury. Hum. Gene Ther. 2010, 21, 713–727. [Google Scholar] [CrossRef]
- Weiss, D.J.; Bates, J.H.T.; Gilbert, T.W.; Liles, W.C.; Lutzko, C.; Rajagopal, J.; Prockop, D. Stem Cells and Cell Therapies in Lung Biology and Diseases: Conference Report. Ann. Am. Thorac. Soc. 2013, 10, S25–S44. [Google Scholar] [CrossRef] [Green Version]
- Gazit, D.; Turgeman, G.; Kelley, P.; Wang, E.; Jalenak, M.; Zilberman, Y.; Moutsatsos, I. Engineered pluripotent mesenchymal cells integrate and differentiate in regenerating bone: A novel cell-mediated gene therapy. J. Gene Med. 1999, 1, 121–133. [Google Scholar] [CrossRef]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.-H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Fiore, E.J.; Bayo, J.M.; Garcia, M.G.; Malvicini, M.; Lloyd, R.; Piccioni, F.; Rizzo, M.; Peixoto, E.; Sola, M.B.; Atorrasagasti, C.; et al. Mesenchymal Stromal Cells Engineered to Produce IGF-I by Recombinant Adenovirus Ameliorate Liver Fibrosis in Mice. Stem Cells Dev. 2014, 24, 791–801. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Advantages and Disadvantages | Citations |
|---|---|---|
| TILs | Patient specific; difficult to obtain; may be highly cancer specific. TCR is typically low affinity. Cells engineered to express IL-2, IL-7, IL-12, IL-15, and IL-18 show enhanced expansion and function; IL-12 secretion produced toxicity. | [11,12,13,14,15,16,17,18] |
| TCR engineered T cells | High affinity and specific; patient specific or disease specific. Difficult to make. Mispairing with endogenous TCR and variability of expression are issues. | [19,20] |
| CAR T cells | Highly effective with B cell neoplasms. Patient specific, expensive, and sometimes toxic. Already an established carrier of many biologics as “armored CARs”. | [6,21] |
| Macrophages | Limited experience to date in secreting drugs. May be difficult to obtain and expand. Can link the innate and adaptive immune response. | [22,23,24] |
| B cells | Cells are capable of large protein production. May be difficult to obtain and expand. | [25,26,27,28,29,30,31] |
| CIK and NK cells | May be less toxic than T cells. Less GVHD and CRS may allow allogeneic uses. IL-15 armoring prolongs activity. | [32,33,34] |
| iPSCs | Off-the-shelf cells are possible. Risks of insertional mutagenesis exist. | [35,36,37,38,39] |
| Vector Design or Approach | Advantages and Disadvantages | Citations |
|---|---|---|
| Multicistronic with 2A peptide or furin cleavage site | Small size. Allows the production of separate proteins from one promotor, but cistrons cannot be differentially regulated. Varied expression on either side of the 2A element. | [74,75] |
| Multiple promoters or bicistronic with an IRES site | Large size and often reduced expression of the second product, which may or may not be desired. | [78,79] |
| Co-transduction | Efficiencies often low and increases cost and complexity. | [80,81,82] |
| Nanoparticle with DNA or RNA for in vivo use | Allows off-the-shelf engineering as cells manipulated in vivo. Size of constructs and persistence may be limiting. RNA may require multiple doses. | [83,84,85] |
| Gene editing and transposons. | Allows control of the insertion site, reducing potential adverse effects and controlling expression; reduces TCR misparing. Low efficiency. | [86,87,88,89] |
| Implanted polymers for in vivo use | Allows off-the-shelf engineering as cells manipulated in vivo. Limited by access to tumors. Long-term effects of implant unknown. | [90,91,92] |
| Gating System | System Type | Logic Decision Made by Cell (Examples) | Citations |
|---|---|---|---|
| Autonomous Gating Systems | Canonical CAR T cell | If antigen, then activate to kill cells. | [174] |
| Multiantigen activation | If multiple antigens, then activate to kill cells. | [175] | |
| Activation dependent | If activated, then initiate transcription of transgene. | [176] | |
| SynNotch | If membrane-bound ligand, then initiate transcription of transgene | [166,177,178] | |
| MESA | If soluble ligand, then initiate transcription of transgene. | [179] | |
| TME gated | If TME, then do B. | [180,181] | |
| iCAR | If off-target ligand, then do not kill cell antigen-positive cells. | [182] | |
| Remote-controlled Gating Systems | Kill switch | If drug is present, then end therapy. | [183,184] |
| SUPRA CAR and UniCAR | If modular recognition molecule bound to cells, then kill cells. | [185,186] | |
| Synthetic receptor/ligand pairs | If drug, then do or do not B. | [187,188,189] | |
| Geography restricted | If localized external stimulus, then do B. | [190,191] |
| Characteristic | Systemic Drug Delivery | Targeted Cellular Micropharmacy (TCM) |
|---|---|---|
| Oral or subcutaneous administration | Yes, for many agents. | No. Generally intravenous. |
| Off-the-shelf availability; Long-term storage | Yes, for most agents | Generally, not currently, but methods to change this are in development. |
| Control of doses and schedule | Yes, but dose at target site can be variable | Limited to cases in which gating or prodrugs are used. |
| Systemic toxicity | Often. Can be severe or fatal. | Promises to create less systemic toxicity for the cell-delivered drug. |
| Therapeutic index (TI) | Often quite limited, resulting in poor efficacy and systemic toxicity. | Local expansion promises to improve TI. Allows drug secretion at target site only. |
| “Logic” or feedback driven actions | No. | “Smart” logic gates engineered into some forms. |
| Persistence in body | Typically, hours to days. | Can be days or months to years. |
| Reactivation when needed. | No. | Yes. |
| Cost | Low to high. | Currently extremely high. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardner, T.J.; Bourne, C.M.; Dacek, M.M.; Kurtz, K.; Malviya, M.; Peraro, L.; Silberman, P.C.; Vogt, K.C.; Unti, M.J.; Brentjens, R.; et al. Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers 2020, 12, 2175. https://doi.org/10.3390/cancers12082175
Gardner TJ, Bourne CM, Dacek MM, Kurtz K, Malviya M, Peraro L, Silberman PC, Vogt KC, Unti MJ, Brentjens R, et al. Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers. 2020; 12(8):2175. https://doi.org/10.3390/cancers12082175
Chicago/Turabian StyleGardner, Thomas J., Christopher M. Bourne, Megan M. Dacek, Keifer Kurtz, Manish Malviya, Leila Peraro, Pedro C. Silberman, Kristen C. Vogt, Mildred J. Unti, Renier Brentjens, and et al. 2020. "Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery" Cancers 12, no. 8: 2175. https://doi.org/10.3390/cancers12082175
APA StyleGardner, T. J., Bourne, C. M., Dacek, M. M., Kurtz, K., Malviya, M., Peraro, L., Silberman, P. C., Vogt, K. C., Unti, M. J., Brentjens, R., & Scheinberg, D. (2020). Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers, 12(8), 2175. https://doi.org/10.3390/cancers12082175