Profound Reprogramming towards Stemness in Pancreatic Cancer Cells as Adaptation to AKT Inhibition

 ,
,  , , , ,
, , , ,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
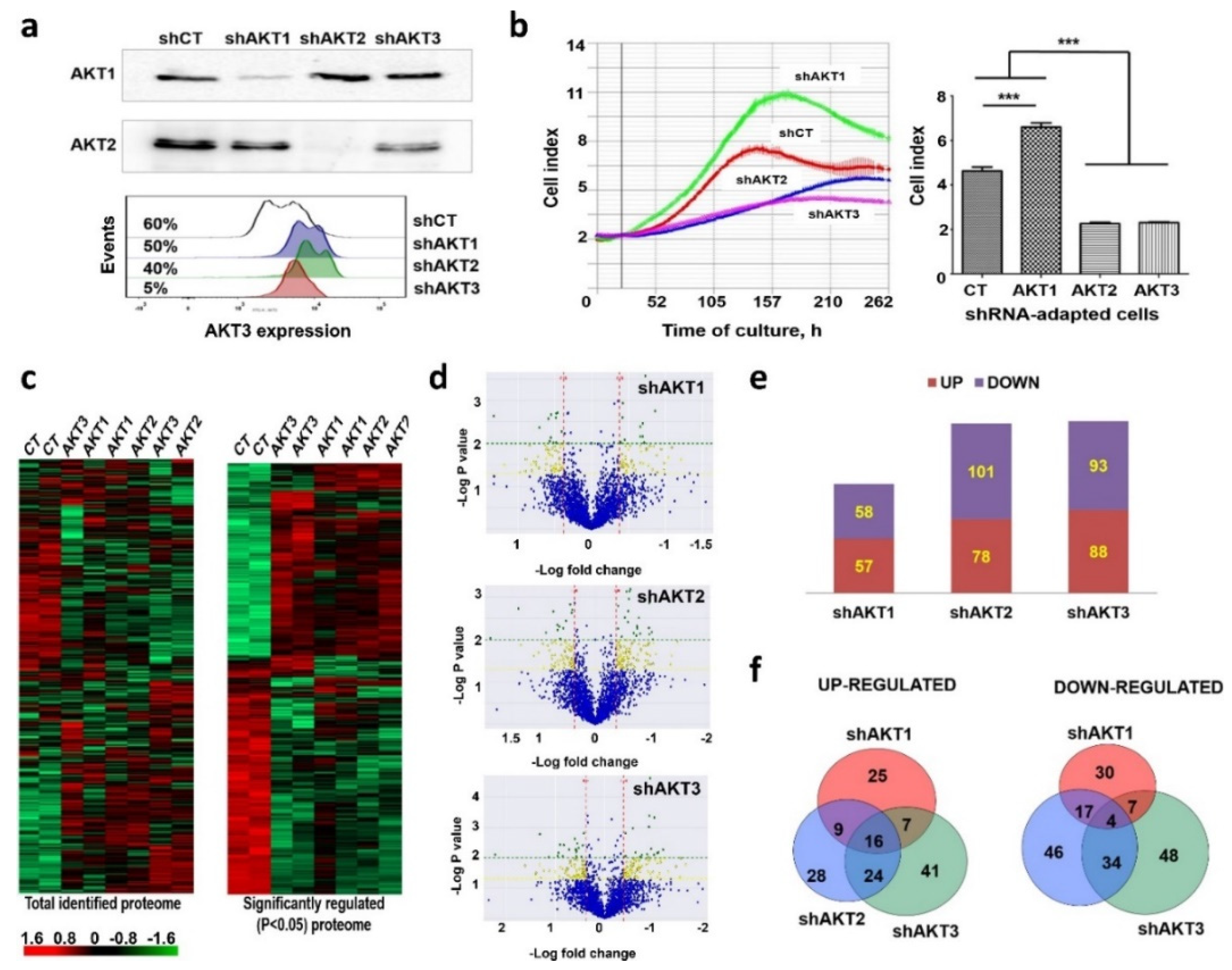
2.1. Adaptation of Cancer Cells to Silencing of AKT Isoforms Triggers Major Changes in Their Proteome
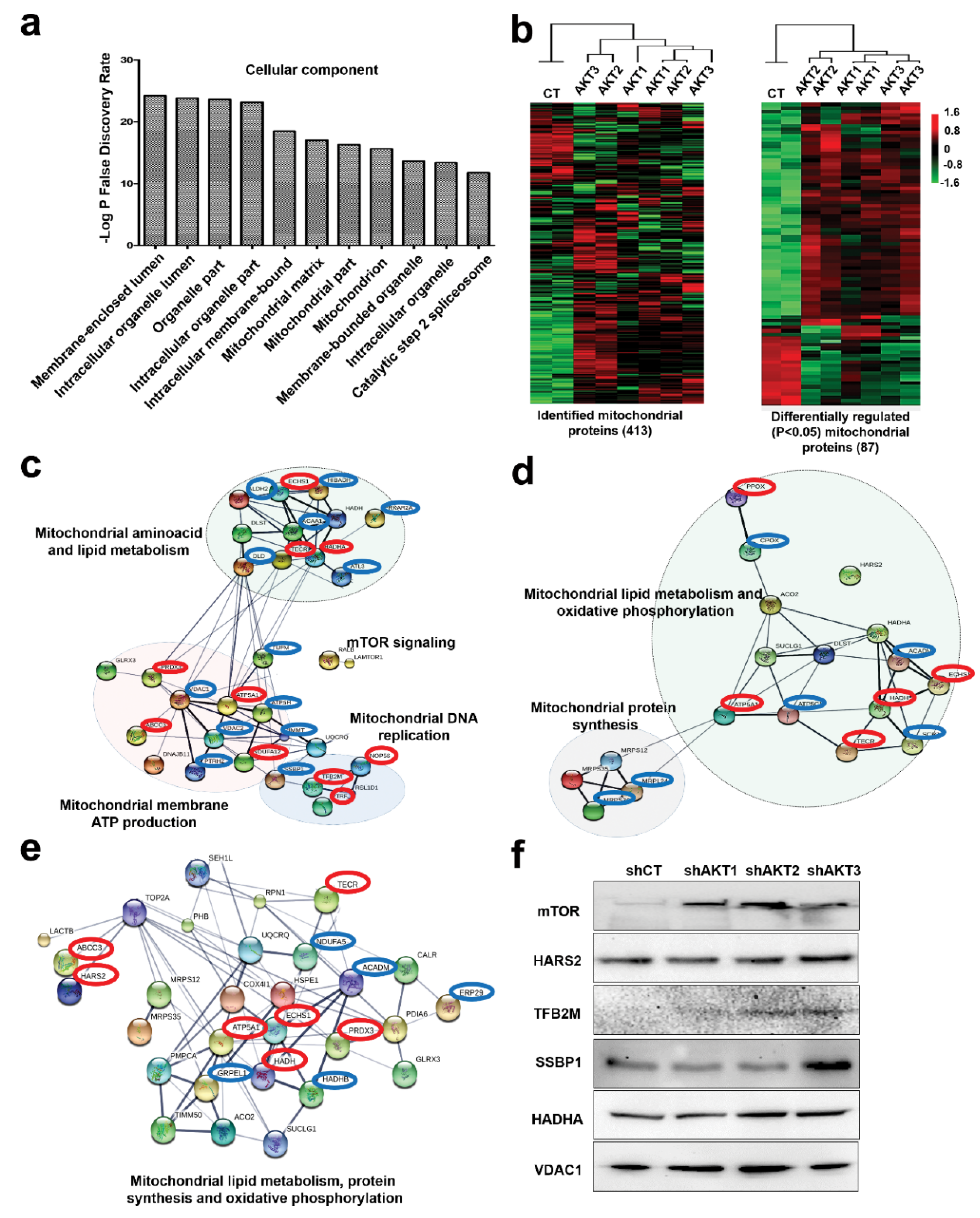
2.2. Adaptation of Cancer Cells to Silencing of AKT Isoforms Causes Profound Mitochondrial Alterations
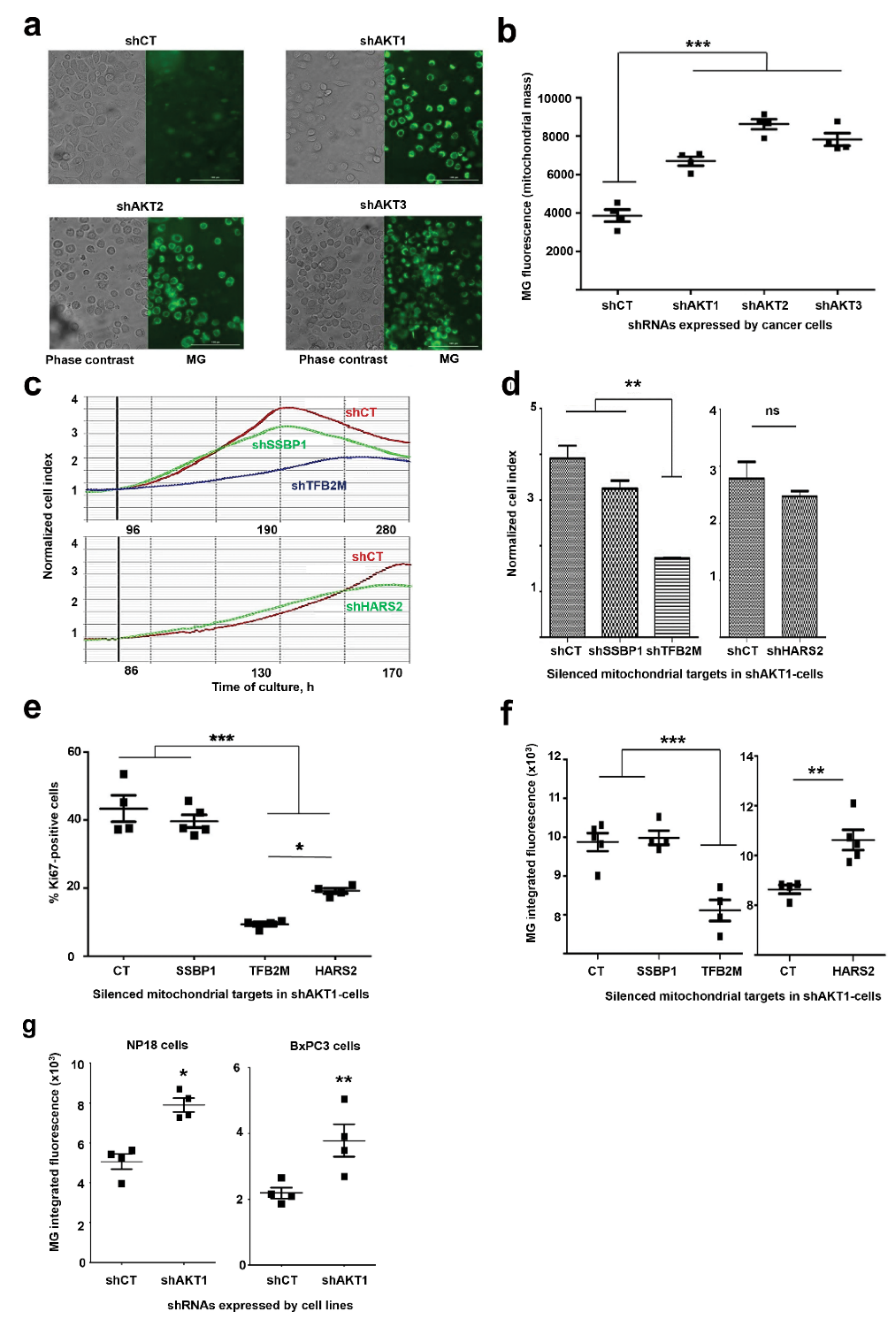
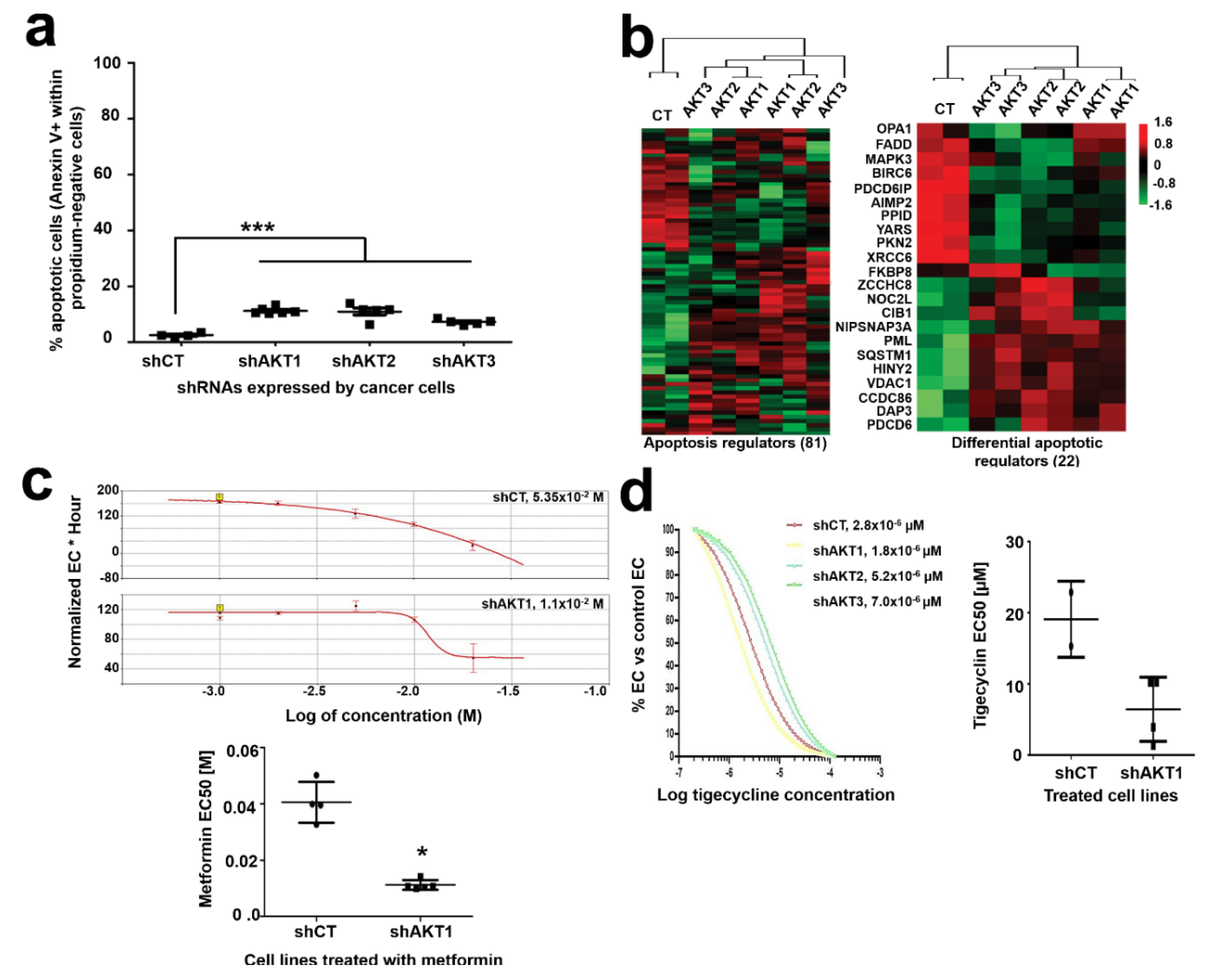
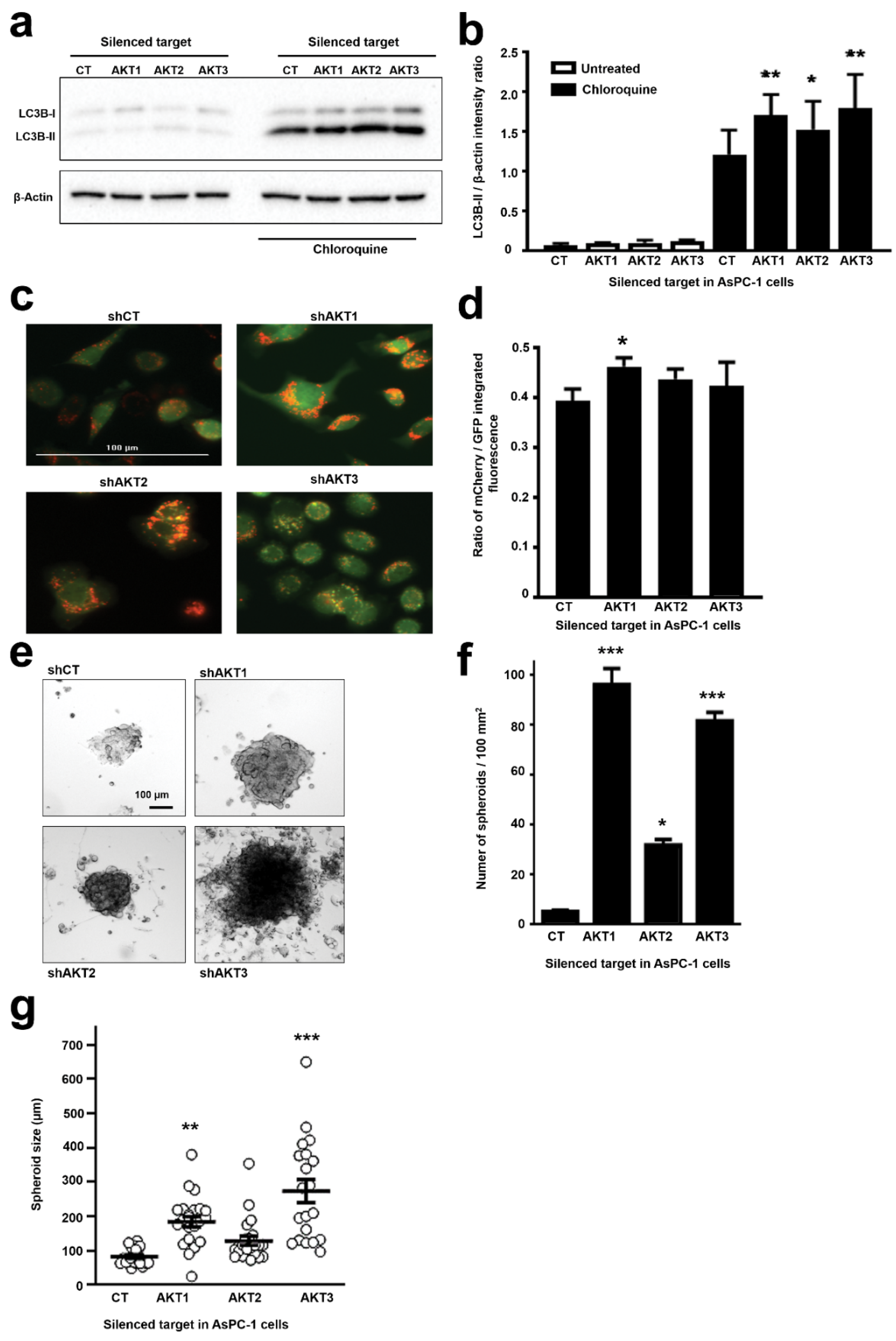
2.3. Adaptation to AKT Silencing Sensitizes Pancreatic Cancer Cells to Mitochondrial Disrupting Agents
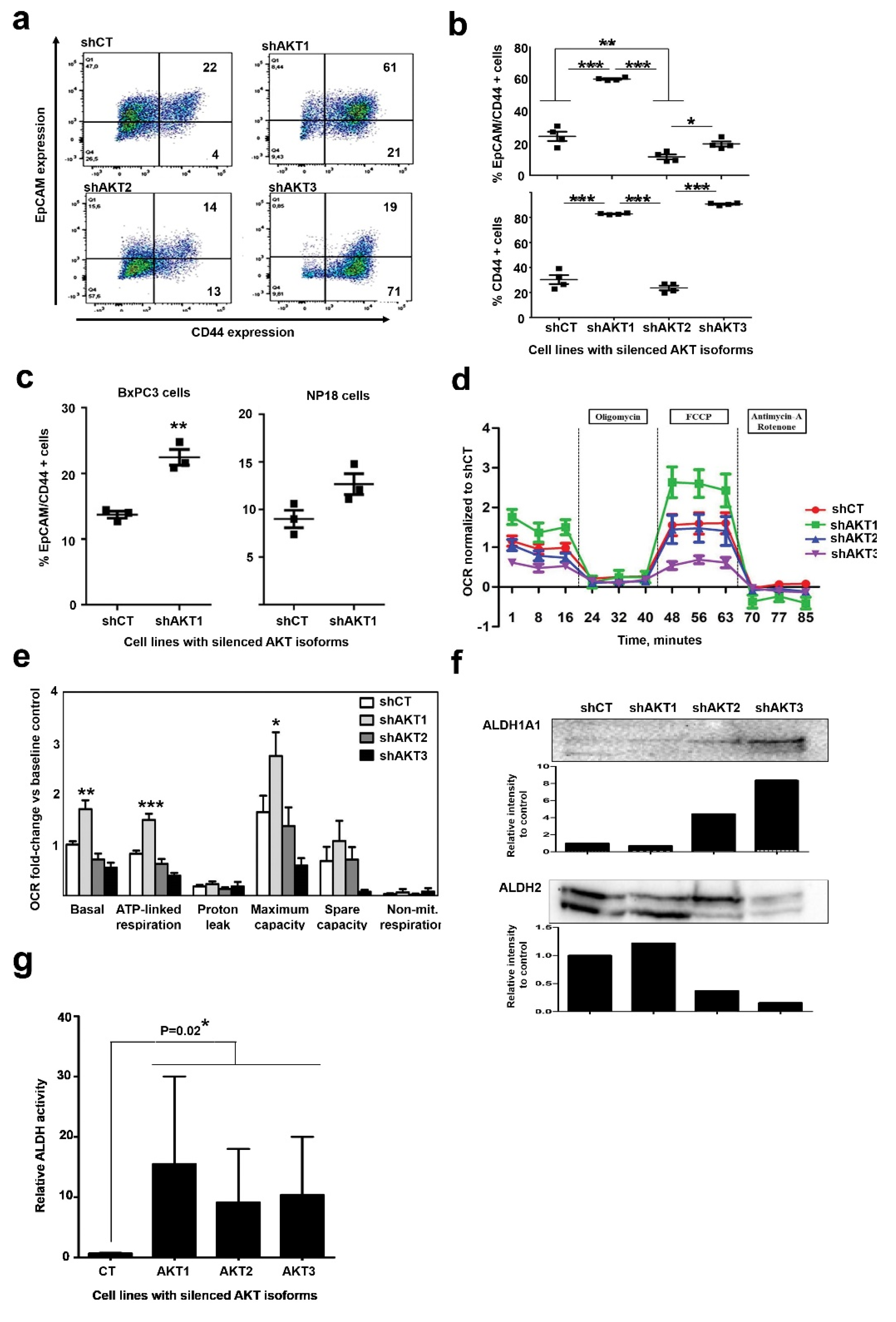
2.4. Adaptation to AKT1 Silencing Causes Acquisition of Cancer Stem Cell Characteristics
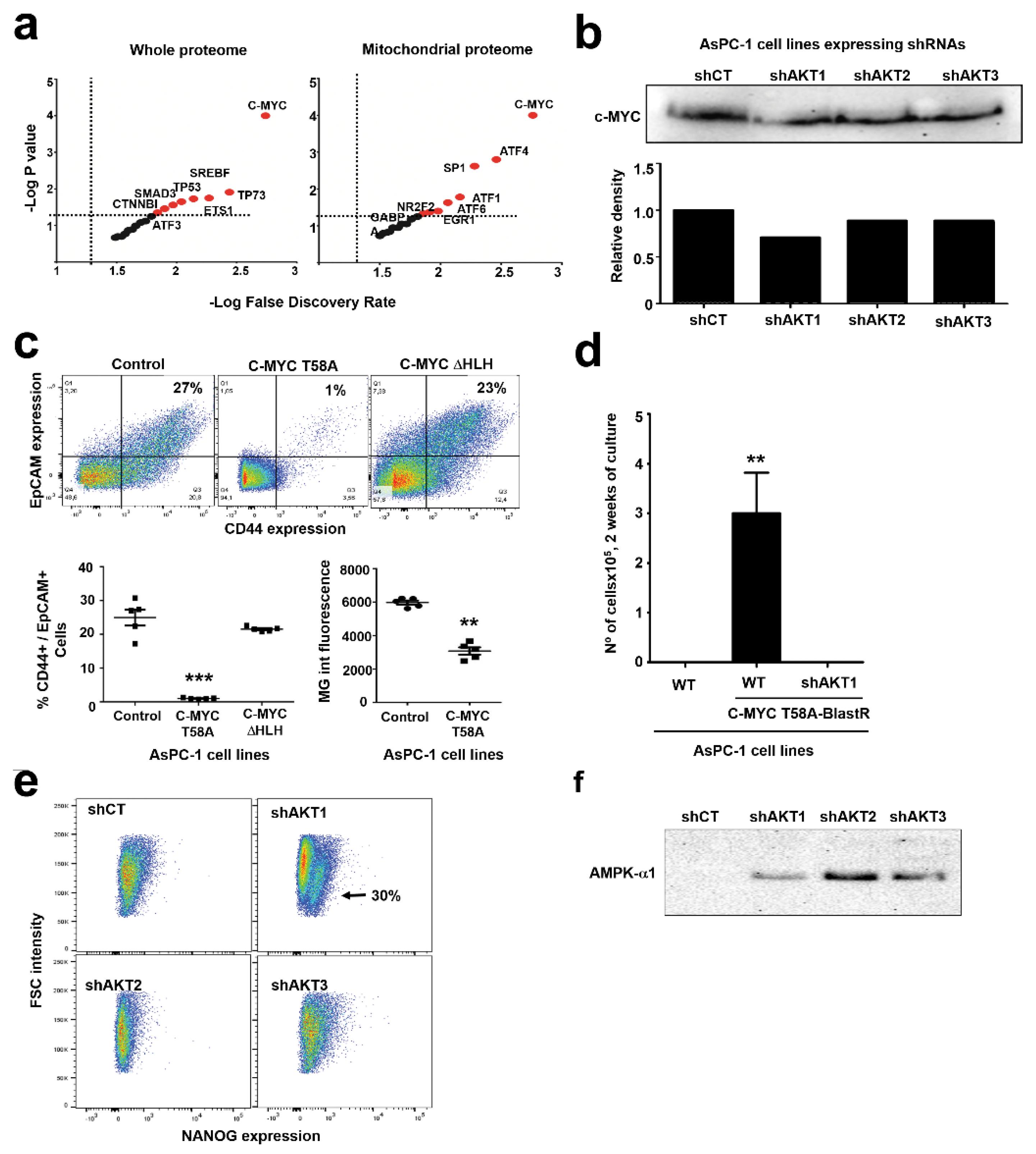
2.5. Adaptation to AKT1 Silencing Uncovers Specific Regulation of C-MYC and NANOG
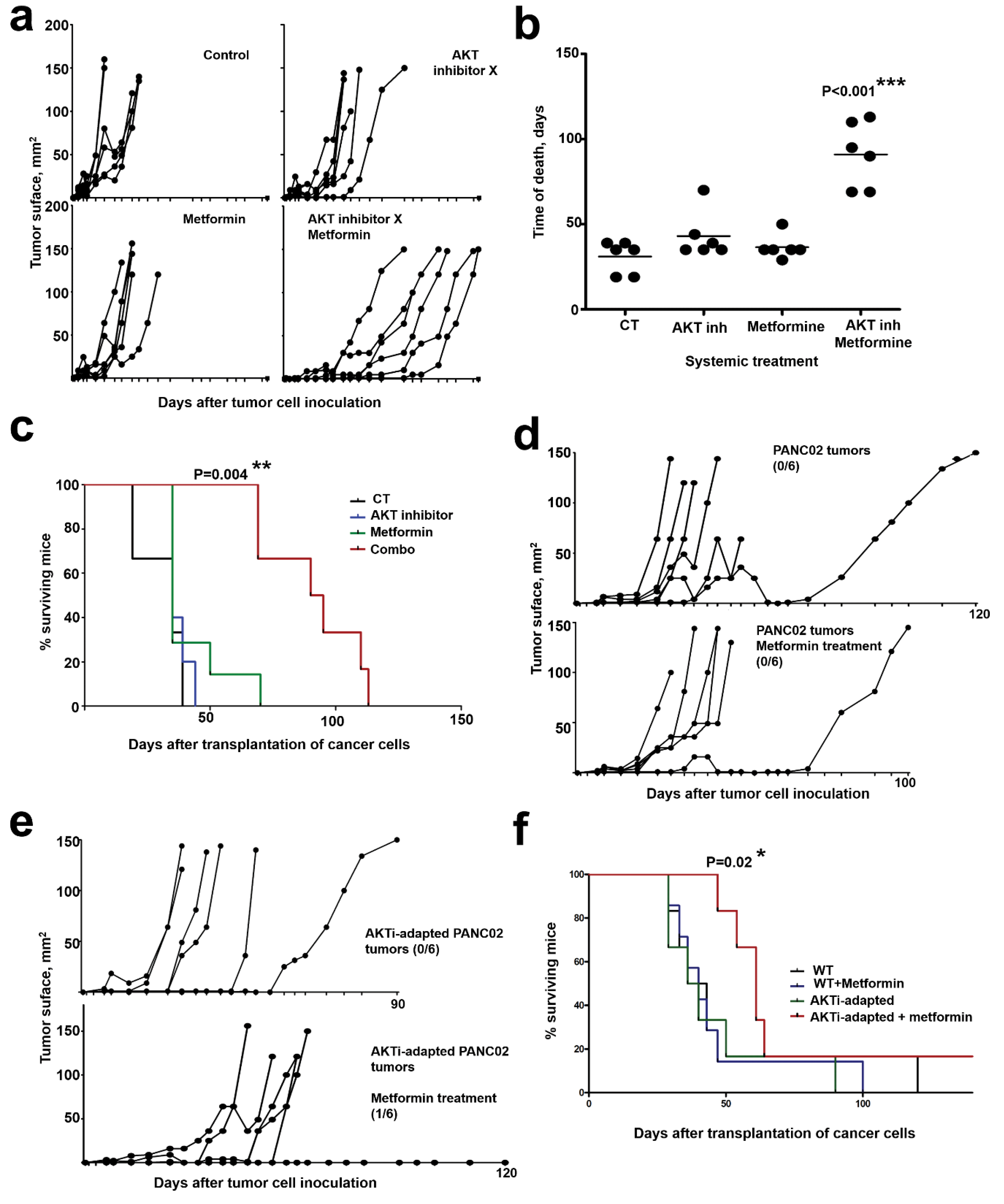
2.6. In Vivo AKT Inhibition Together with Metformin Treatment Increases Therapeutic Efficacy
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Lentivector Construction, Production and Cell Transduction
4.3. Mitochondrial Staining and Respiration
4.4. Autophagy
4.5. Immunoblot
4.6. Flow Cytometry
4.7. Proteomics
4.8. Data Analysis
4.9. Bioinformatic Analyses
4.10. In Vivo Experiments
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Soares, H.P.; Ni, Y.; Kisfalvi, K.; Sinnett-Smith, J.; Rozengurt, E. Different Patterns of Akt and ERK Feedback Activation in Response to Rapamycin, Active-Site mTOR Inhibitors and Metformin in Pancreatic Cancer Cells. PLoS ONE 2013, 8, e57289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iriana, S.; Ahmed, S.; Gong, J.; Annamalai, A.A.; Tuli, R.; Hendifar, A.E. Targeting mTOR in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2016, 6, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, H.L.; Wroblewski, K.; Wallace, J.A.; Hall, M.J.; Locker, G.; Nattam, S.; Agamah, E.; Stadler, W.M.; Vokes, E.E. Gemcitabine Plus Sorafenib in Patients with Advanced Pancreatic Cancer: A Phase II Trial of the University of Chicago Phase II Consortium. Invest. New Drugs 2012, 30, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Klemm, F.; Joyce, J.A. Microenvironmental Regulation of Therapeutic Response in Cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and Cancer Chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Linnerth-Petrik, N.M.; Santry, L.A.; Petrik, J.J.; Wootton, S.K. Opposing Functions of Akt Isoforms in Lung Tumor Initiation and Progression. PLoS ONE 2014, 9, e94595. [Google Scholar] [CrossRef] [Green Version]
- Hollander, M.C.; Maier, C.R.; Hobbs, E.A.; Ashmore, A.R.; Linnoila, R.I.; Dennis, P.A. Akt1 Deletion Prevents Lung Tumorigenesis by Mutant K-ras. Oncogene 2011, 30, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Yoeli-Lerner, M.; Yiu, G.K.; Rabinovitz, I.; Erhardt, P.; Jauliac, S.; Toker, A. Akt Blocks Breast Cancer Cell Motility and Invasion through the Transcription Factor NFAT. Mol. Cell 2005, 20, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Virtakoivu, R.; Pellinen, T.; Rantala, J.K.; Perala, M.; Ivaska, J. Distinct Roles of AKT Isoforms in Regulating Beta1–Integrin Activity, Migration, and Invasion in Prostate Cancer. Mol. Biol. Cell 2012, 23, 3357–3369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist Transcriptionally Up-Regulates AKT2 in Breast Cancer Cells Leading to Increased Migration, Invasion, and Resistance to Paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 Signaling in Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.U.; Sangai, T.; Akcakanat, A.; Chen, H.; Wei, C.; Meric-Bernstam, F. Vertical Inhibition of the PI3K/Akt/mTOR Pathway is Synergistic in Breast Cancer. Oncogenesis 2017, 6, e385. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR Coordinates Protein Synthesis, Mitochondrial Activity and Proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Jurado, S.; Diaz-Colunga, J.; das Neves, R.P.; Martinez-Lorente, A.; Almazan, F.; Guantes, R.; Iborra, F.J. Mitochondrial Levels Determine Variability in Cell Death by Modulating Apoptotic Gene Expression. Nat. Commun. 2018, 9, 389. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Jhas, B.; Sriskanthadevan, S.; Skrtic, M.; Sukhai, M.A.; Voisin, V.; Jitkova, Y.; Gronda, M.; Hurren, R.; Laister, R.C.; Bader, G.D.; et al. Metabolic Adaptation to Chronic Inhibition of Mitochondrial Protein Synthesis in Acute Myeloid Leukemia cells. PLoS ONE 2013, 8, e58367. [Google Scholar] [CrossRef]
- Heiler, S.; Wang, Z.; Zoller, M. Pancreatic Cancer Stem Cell Markers and Exosomes—the Incentive Push. World J. Gastroenterol. 2016, 22, 5971–6007. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde Dehydrogenase 1A1 in Stem Cells and Cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Mo, Y.; Li, M.T.; Zou, S.W.; Cheng, Z.L.; Sun, Y.P.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. NOTCH-Induced Aldehyde Dehydrogenase 1A1 Deacetylation Promotes Breast Cancer Stem Cells. J. Clin. Investig. 2014, 124, 5453–5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.G.; Macleod, K.F. Autophagy, Cancer Stem Cells and Drug Resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, S.; Martin-Hijano, L.; Alcala, S.; Alonso-Nocelo, M.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd ed.). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Noel, P.; Munoz, R.; Rogers, G.W.; Neilson, A.; Von Hoff, D.D.; Han, H. Preparation and Metabolic Assay of 3-dimensional Spheroid Co-cultures of Pancreatic Cancer Cells and Fibroblasts. J. Vis. Exp. 2017, 126, e56081. [Google Scholar] [CrossRef] [Green Version]
- Thibodeaux, C.A.; Liu, X.; Disbrow, G.L.; Zhang, Y.; Rone, J.D.; Haddad, B.R.; Schlegel, R. Immortalization and Transformation of Human Mammary Epithelial Cells by a Tumor-Derived Myc Mutant. Breast Cancer Res. Treat. 2009, 116, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES Cell Self-Renewal and Pluripotency by a Myc-Dependent Mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Hyslop, L.; Stojkovic, M.; Armstrong, L.; Walter, T.; Stojkovic, P.; Przyborski, S.; Herbert, M.; Murdoch, A.; Strachan, T.; Lako, M. Downregulation of NANOG Induces Differentiation of Human Embryonic stem Cells to Extraembryonic Lineages. Stem. Cells 2005, 23, 1035–1043. [Google Scholar] [CrossRef]
- Jeter, C.R.; Yang, T.; Wang, J.; Chao, H.P.; Tang, D.G. Concise Review: NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions. Stem. Cells 2015, 33, 2381–2390. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, H.; Broxmeyer, H. AMPK Regulates Nanog Gene Expression via p53 in Mouse Embryonic Stem Cells. Blood 2009, 114, 2537. [Google Scholar] [CrossRef]
- Liu, Y.; Yamashita, J.K. AMPK Activators Contribute to Maintain Naive Pluripotency in Mouse Embryonic Stem Cells. Biochem. Biophys. Res. Commun. 2019, 509, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Partecke, L.I.; Sendler, M.; Kaeding, A.; Weiss, F.U.; Mayerle, J.; Dummer, A.; Nguyen, T.D.; Albers, N.; Speerforck, S.; Lerch, M.M.; et al. A Syngeneic Orthotopic Murine Model of Pancreatic Adenocarcinoma in the C57/BL6 Mouse Using the Panc02 and 6606PDA Cell Lines. Eur. Surg. Res. 2011, 47, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Malkomes, P.; Lunger, I.; Luetticke, A.; Oppermann, E.; Haetscher, N.; Serve, H.; Holzer, K.; Bechstein, W.O.; Rieger, M.A. Selective AKT Inhibition by MK-2206 Represses Colorectal Cancer-Initiating Stem Cells. Ann. Surg. Oncol. 2016, 23, 2849–2857. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.K.; Paul, R.; Haas, M.; Wang, C.; Guan, J.L. Improved Efficacy of Mitochondrial Disrupting Agents upon Inhibition of Autophagy in a Mouse Model of BRCA1-Deficient Breast Cancer. Autophagy 2018, 14, 1214–1225. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer Stem Cells: An Evolving Concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR Signaling Pathway in Cancer Stem Cells: From Basic Research to Clinical Application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Noh, K.H.; Kim, B.W.; Song, K.H.; Cho, H.; Lee, Y.H.; Kim, J.H.; Chung, J.Y.; Kim, J.H.; Hewitt, S.M.; Seong, S.Y.; et al. Nanog Signaling in Cancer Promotes Stem-Like Phenotype and Immune Evasion. J. Clin. Investig. 2012, 122, 4077–4093. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yu, W.N.; Chen, X.; Peng, X.D.; Jeon, S.M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Akt Isoforms and Glucose Homeostasis—the Leptin Connection. Trends Endocrinol. Metab. 2011, 22, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that Target Mitochondria Effectively Eradicate Cancer Stem Cells, across Multiple Tumor Types: Treating Cancer Like an Infectious Disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.M.; Koorstra, J.B.; Rajeshkumar, N.V.; He, X.; et al. Prognostic Significance of Tumorigenic Cells with Mesenchymal Features in Pancreatic Adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem. Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde Dehydrogenase: Its Role as a Cancer Stem Cell Marker Comes down to the Specific Isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef]
- Yan, S.; Wu, G. Could ALDH2(*)2 be the Reason for Low Incidence and Mortality of Ovarian Cancer for East Asia Women? Oncotarget 2018, 9, 12503–12512. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is Activated in Pancreatic Cancer Cells and Correlates with Poor Patient Outcome. Cancer Sci. 2008, 99, 1813–1819. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Martinez de Morentin, X.; Blanco-Luquin, I.; Fernandez-Irigoyen, J.; Zudaire, I.; Liechtenstein, T.; Arasanz, H.; Lozano, T.; Casares, N.; Chaikuad, A.; et al. A Core of Kinase-Regulated Interactomes Defines the Neoplastic MDSC Lineage. Oncotarget 2015, 6, 27160–27175. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr. Protoc. Toxicol. 2014, 60, 25.2.1–25.2.16. [Google Scholar] [CrossRef]
- Zuazo, M.; Arasanz, H.; Fernandez-Hinojal, G.; Garcia-Granda, M.J.; Gato, M.; Bocanegra, A.; Martinez, M.; Hernandez, B.; Teijeira, L.; Morilla, I.; et al. Functional Systemic CD4 Immunity is Required for Clinical Responses to PD-L1/PD-1 Blockade Therapy. EMBO Mol. Med. 2019, 11, e10293. [Google Scholar] [CrossRef] [PubMed]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous Analysis of Relative Protein Expression Levels across Multiple Samples Using iTRAQ Isobaric Tags with 2D Nano LC-MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The Paragon Algorithm, a Next Generation Search Engine that Uses Sequence Temperature Values and Feature Probabilities to Identify Peptides from Tandem Mass Spectra. Mol. Cell Proteomics 2007, 6, 1638–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liechtenstein, T.; Perez-Janices, N.; Gato, M.; Caliendo, F.; Kochan, G.; Blanco-Luquin, I.; Van der Jeught, K.; Arce, F.; Guerrero-Setas, D.; Fernandez-Irigoyen, J.; et al. A Highly Efficient Tumor-Infiltrating MDSC Differentiation System for Discovery of Anti-Neoplastic Targets, which Circumvents the Need for Tumor Establishment in Mice. Oncotarget 2014, 5, 7843–7857. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Shilov, I.V.; Seymour, S.L. Nonlinear Fitting Method for Determining Local False Discovery Rates from Decoy Database Searches. J. Proteome Res. 2008, 7, 3661–3667. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arasanz, H.; Hernández, C.; Bocanegra, A.; Chocarro, L.; Zuazo, M.; Gato, M.; Ausin, K.; Santamaría, E.; Fernández-Irigoyen, J.; Fernandez, G.; et al. Profound Reprogramming towards Stemness in Pancreatic Cancer Cells as Adaptation to AKT Inhibition. Cancers 2020, 12, 2181. https://doi.org/10.3390/cancers12082181
Arasanz H, Hernández C, Bocanegra A, Chocarro L, Zuazo M, Gato M, Ausin K, Santamaría E, Fernández-Irigoyen J, Fernandez G, et al. Profound Reprogramming towards Stemness in Pancreatic Cancer Cells as Adaptation to AKT Inhibition. Cancers. 2020; 12(8):2181. https://doi.org/10.3390/cancers12082181
Chicago/Turabian StyleArasanz, Hugo, Carlos Hernández, Ana Bocanegra, Luisa Chocarro, Miren Zuazo, Maria Gato, Karina Ausin, Enrique Santamaría, Joaquín Fernández-Irigoyen, Gonzalo Fernandez, and et al. 2020. "Profound Reprogramming towards Stemness in Pancreatic Cancer Cells as Adaptation to AKT Inhibition" Cancers 12, no. 8: 2181. https://doi.org/10.3390/cancers12082181
APA StyleArasanz, H., Hernández, C., Bocanegra, A., Chocarro, L., Zuazo, M., Gato, M., Ausin, K., Santamaría, E., Fernández-Irigoyen, J., Fernandez, G., Santamaria, E., Rodríguez, C., Blanco-Luquin, I., Vera, R., Escors, D., & Kochan, G. (2020). Profound Reprogramming towards Stemness in Pancreatic Cancer Cells as Adaptation to AKT Inhibition. Cancers, 12(8), 2181. https://doi.org/10.3390/cancers12082181