C3G Is Upregulated in Hepatocarcinoma, Contributing to Tumor Growth and Progression and to HGF/MET Pathway Activation

 ,
, 
 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
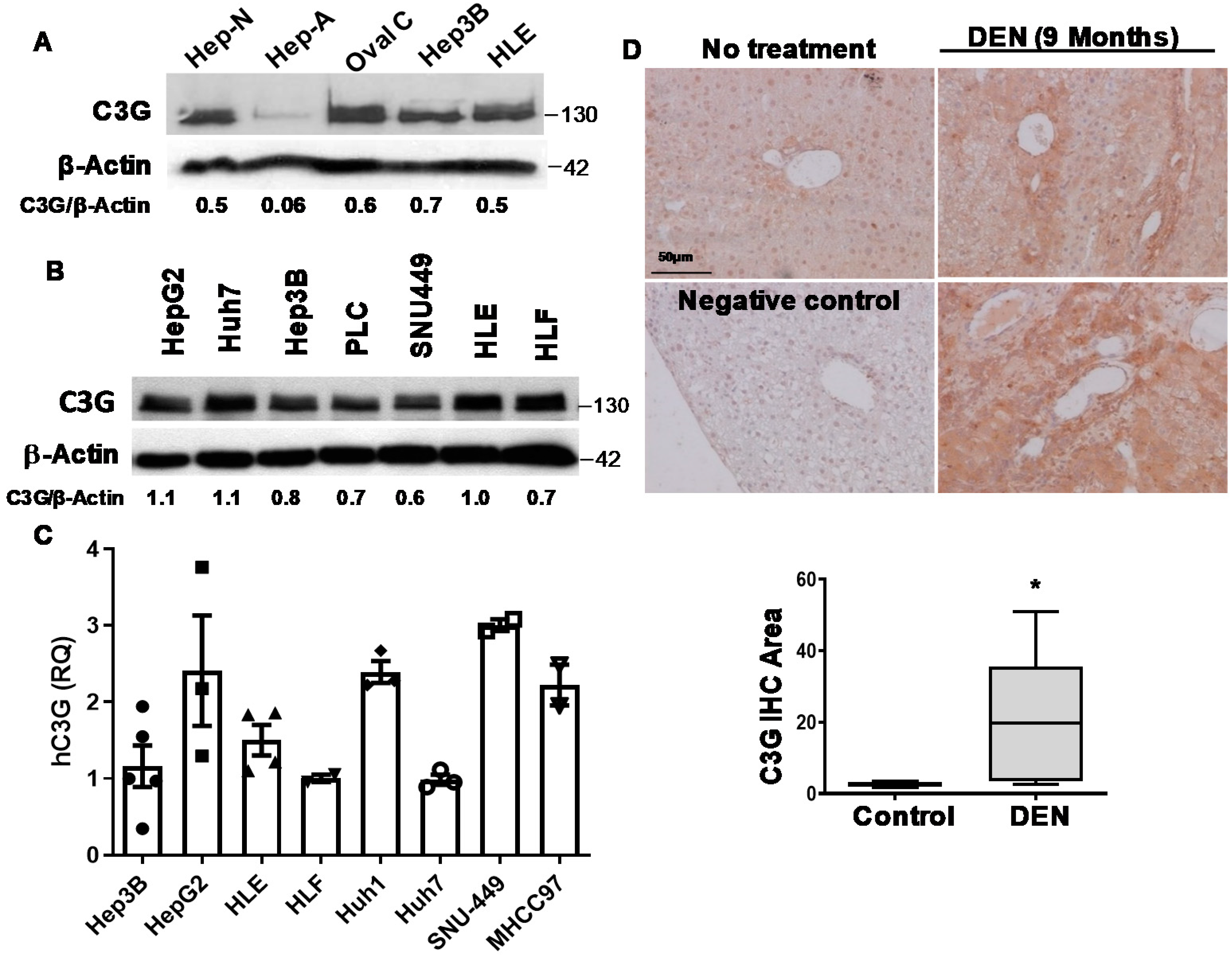
2.1. C3G Is Overexpressed in Mouse and Human HCC
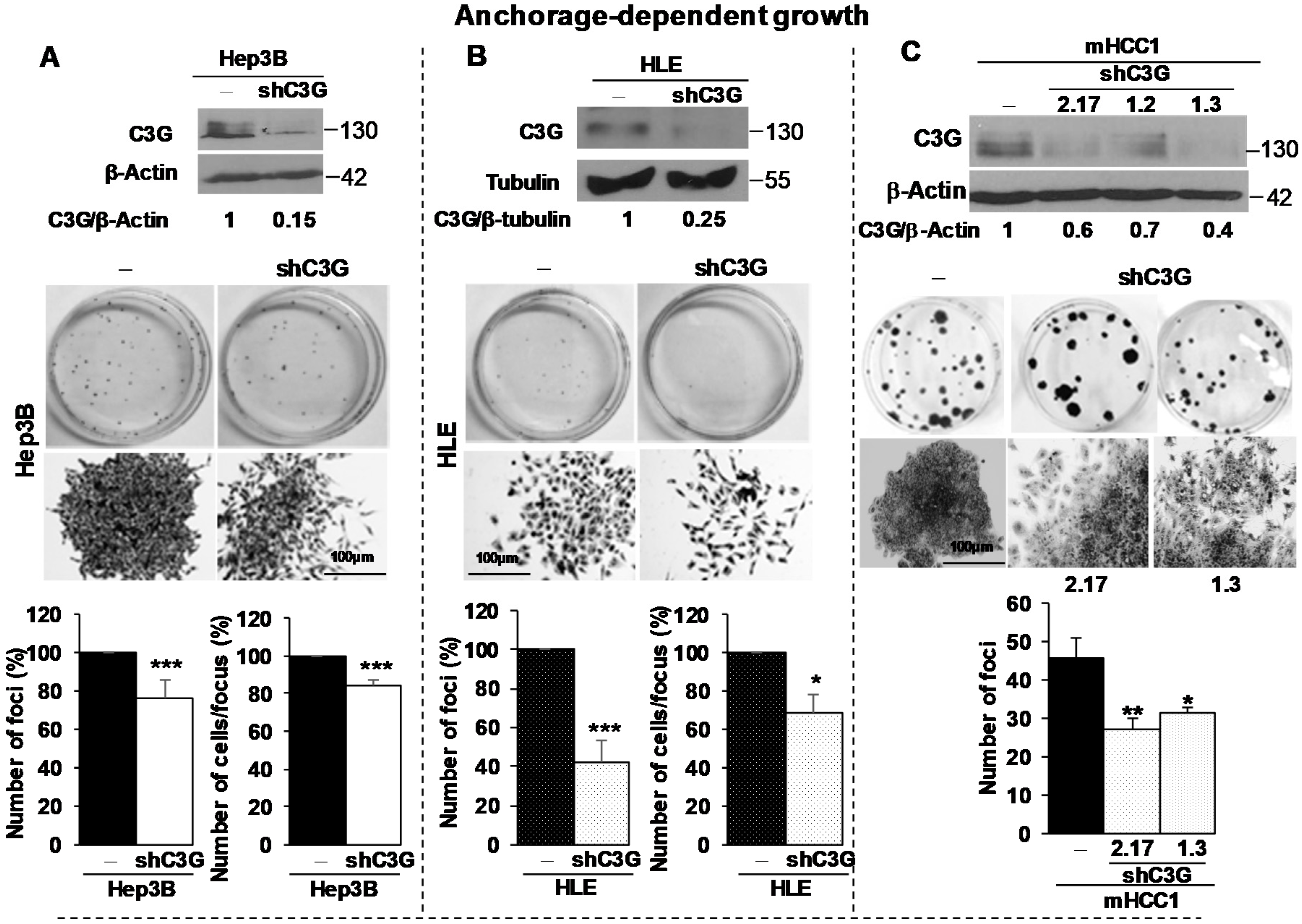
2.2. C3G down-Regulation Reduces Foci Formation by HCC Cells
2.3. C3G Down-Regulation Enhances Migratory and Invasive Properties of HCC Cells by Switching from an Epithelial to a Mesenchymal Phenotype
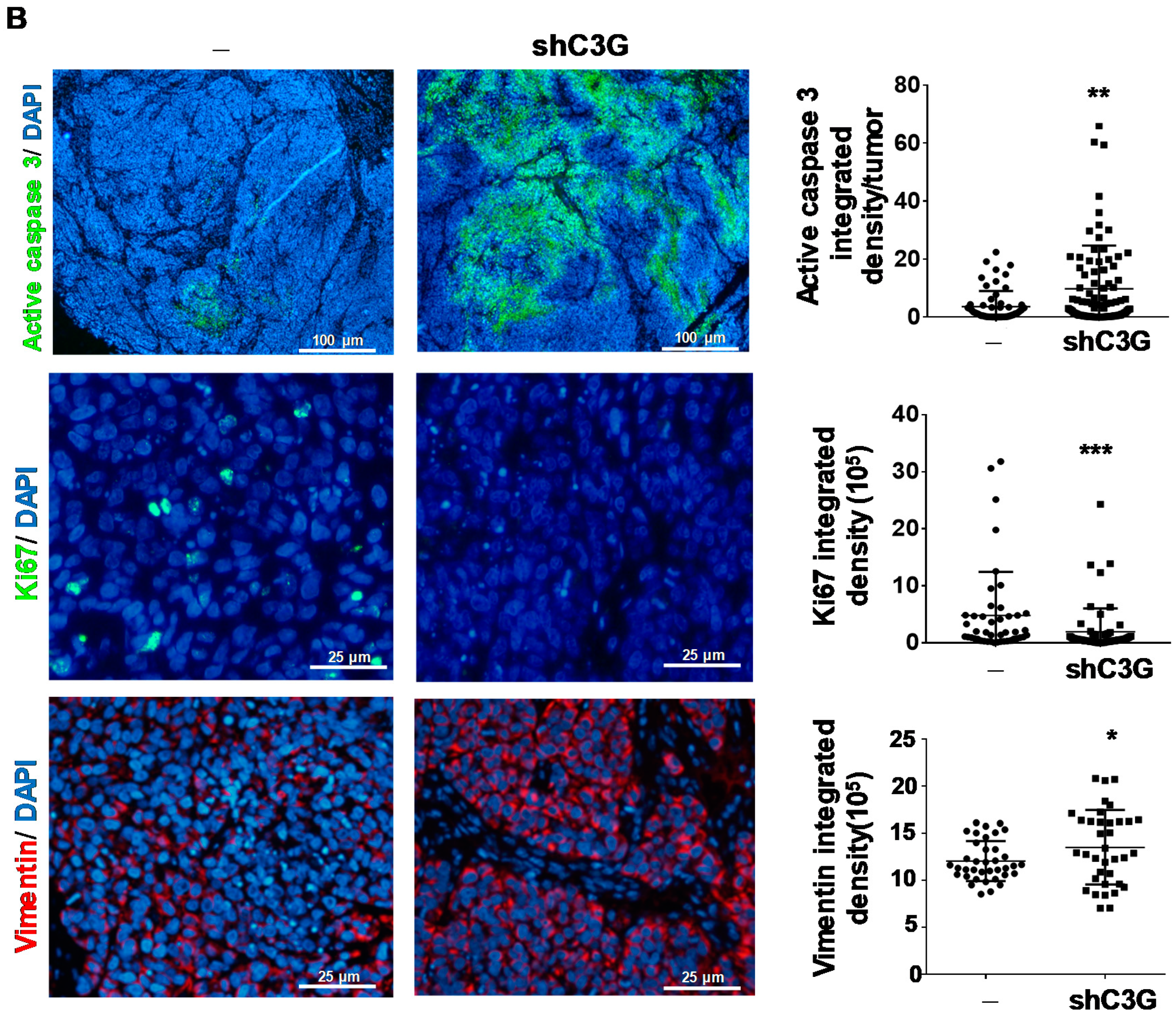
2.4. C3G down-Regulation Reduces Primary HCC Tumor Growth
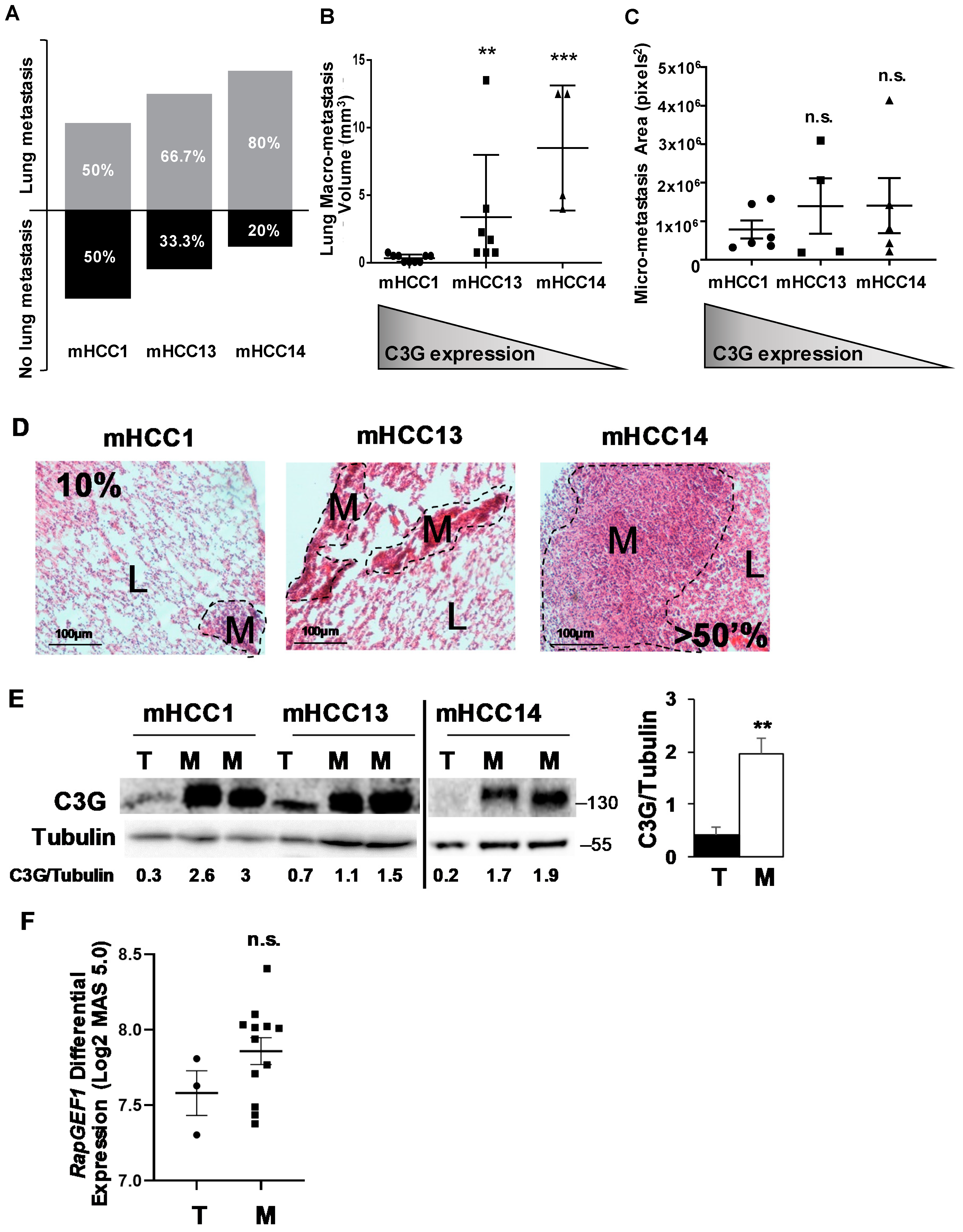
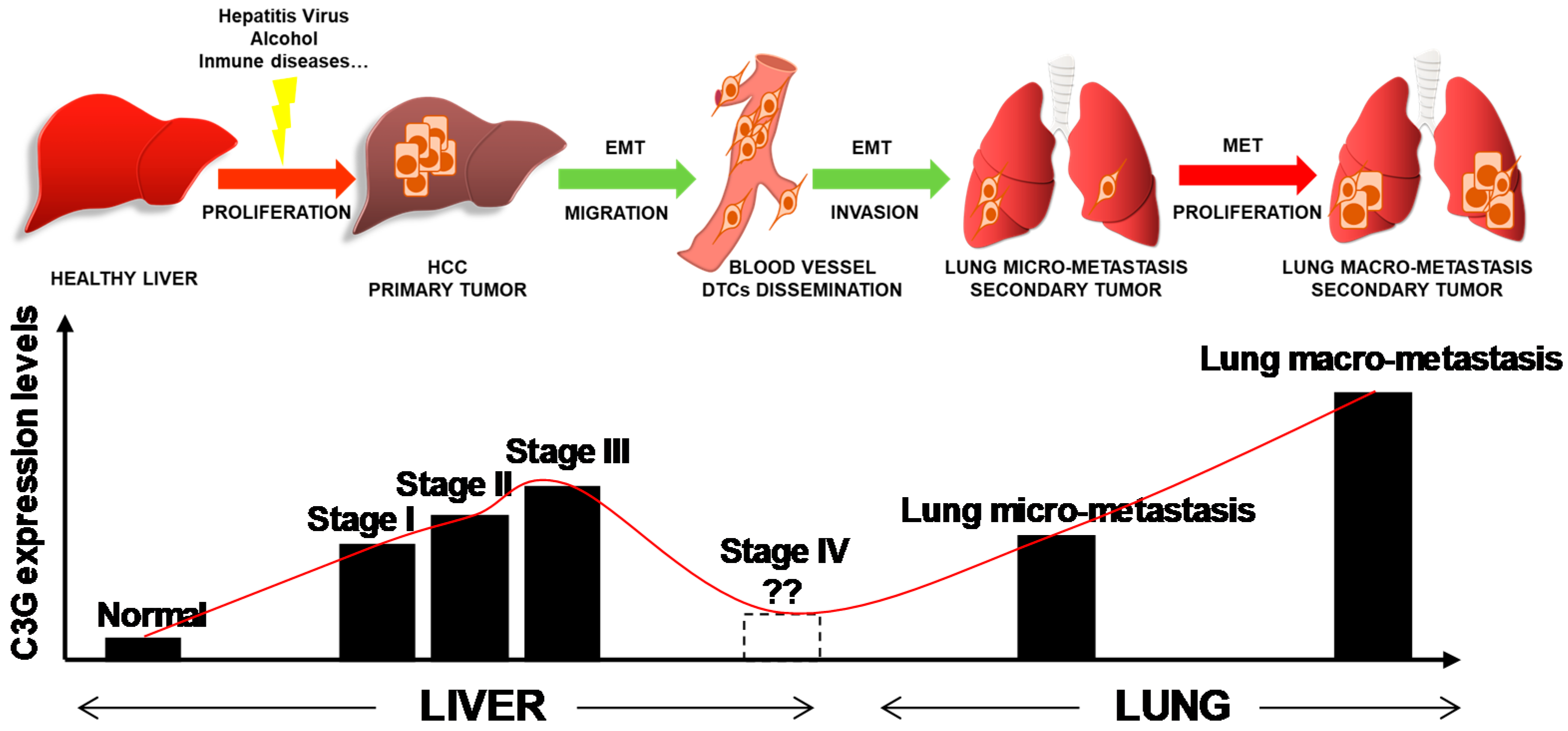
2.5. C3G Levels Inversely Correlate with Lung Metastasis Formation by Mouse Alb-R26Met HCC Cells with Enhanced MET Expression
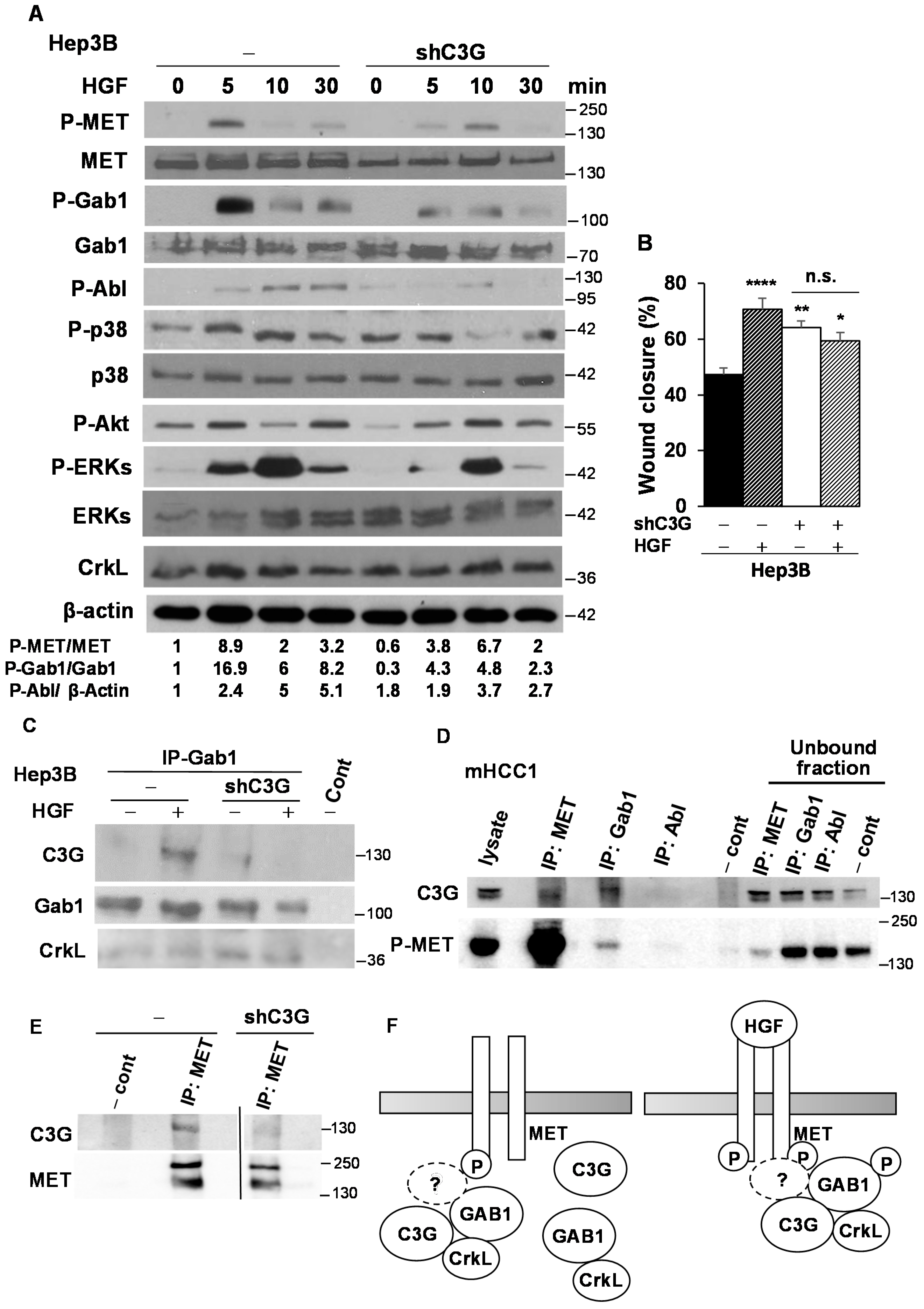
2.6. C3G Ensures Full Activation of the HGF/MET Signaling Pathway in Human HCC Cells
3. Discussion
4. Materials and Methods
4.1. Public Genomic Databases
4.2. Cell Lines and C3G Silencing
4.3. Cell and Tissue Extracts Preparation and Western Blot Analysis
4.4. Immunoprecipitation Assay
4.5. Pull-Down Assay
4.6. Proliferation and Apoptosis Analysis by Flow Cytometry
4.7. Cell Adhesion Assay
4.8. Wound Healing Assay
4.9. Invasion Assay
4.10. Anchorage-Dependent Growth
4.11. Anchorage-Independent Growth
4.12. Induction of EMT with TGFβ
4.13. RT-qPCR Analysis
4.14. Xenograft Assays
4.15. Generation of Lung Metastasis in Mice Using the Alb-R26Met HCC Cell Lines
4.16. DEN-Induced Liver Tumors
4.17. Tissue Samples Staining and Analysis
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Ackowledgements
Funding
Conflicts of Interest
References
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and future treatment of hepatocellular carcinoma: An updated comprehensive review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfuso, B.; El-Khobar, K.E.; Sukowati, C.H.; Tiribelli, C. The multiple origin of cancer stem cells in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. 1), S92–S97. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boix, L.; Rosa, J.L.; Ventura, F.; Castells, A.; Bruix, J.; Rodes, J.; Bartrons, R. C-met mRNA overexpression in human hepatocellular carcinoma. Hepatology 1994, 19, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Kaposi-Novak, P.; Lee, J.S.; Gomez-Quiroz, L.; Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Investig. 2006, 116, 1582–1595. [Google Scholar] [CrossRef]
- Furlan, A.; Stagni, V.; Hussain, A.; Richelme, S.; Conti, F.; Prodosmo, A.; Destro, A.; Roncalli, M.; Barila, D.; Maina, F. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death Differ. 2011, 18, 1608–1616. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Jang, H.J.; Lee, J. Prognostic value of c-Met overexpression in hepatocellular carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 90351–90357. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [Green Version]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. C-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Hattori, S.; Nakamura, S.; Kitayama, H.; Noda, M.; Takai, Y.; Kaibuchi, K.; Matsui, H.; Hatase, O.; Takahashi, H.; et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol. Cell. Biol. 1995, 15, 6746–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, T.; Niino, Y.; Tokuda, M.; Hatase, O.; Nakamura, S.; Matsuda, M.; Hattori, S. Activation of R-Ras by Ras-guanine nucleotide-releasing factor. J. Biol. Chem. 1997, 272, 18602–18607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohba, Y.; Ikuta, K.; Ogura, A.; Matsuda, J.; Mochizuki, N.; Nagashima, K.; Kurokawa, K.; Mayer, B.J.; Maki, K.; Miyazaki, J.; et al. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001, 20, 3333–3341. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, C.; Fernandez-Medarde, A.; Rojas, J.M.; Font de Mora, J.; Esteban, L.M.; Santos, E. Transformation suppressor activity of C3G is independent of its CDC25-homology domain. Oncogene 1998, 16, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, C.; Martin-Encabo, S.; Fernandez-Medarde, A.; Santos, E. C3G-mediated suppression of oncogene-induced focus formation in fibroblasts involves inhibition of ERK activation, cyclin A expression and alterations of anchorage-independent growth. Oncogene 2004, 23, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Shivakrupa, R.; Radha, V.; Sudhakar, C.; Swarup, G. Physical and functional interaction between Hck tyrosine kinase and guanine nucleotide exchange factor C3G results in apoptosis, which is independent of C3G catalytic domain. J. Biol. Chem. 2003, 278, 52188–52194. [Google Scholar] [CrossRef] [Green Version]
- Shakyawar, D.K.; Dayma, K.; Ramadhas, A.; Varalakshmi, C.; Radha, V. C3G shows regulated nucleocytoplasmic exchange and represses histone modifications associated with euchromatin. Mol. Biol. Cell 2017, 28, 984–995. [Google Scholar] [CrossRef]
- Shakyawar, D.K.; Muralikrishna, B.; Radha, V. C3G dynamically associates with nuclear speckles and regulates mRNA splicing. Mol. Biol. Cell 2018, 29, 1111–1124. [Google Scholar] [CrossRef]
- Radha, V.; Rajanna, A.; Mitra, A.; Rangaraj, N.; Swarup, G. C3G is required for c-Abl-induced filopodia and its overexpression promotes filopodia formation. Exp. Cell Res. 2007, 313, 2476–2492. [Google Scholar] [CrossRef]
- Radha, V.; Mitra, A.; Dayma, K.; Sasikumar, K. Signalling to actin: Role of C3G, a multitasking guanine-nucleotide-exchange factor. Biosci. Rep. 2011, 31, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Maia, V.; Sanz, M.; Gutierrez-Berzal, J.; de Luis, A.; Gutierrez-Uzquiza, A.; Porras, A.; Guerrero, C. C3G silencing enhances STI-571-induced apoptosis in CML cells through p38 MAPK activation, but it antagonizes STI-571 inhibitory effect on survival. Cell Signal. 2009, 21, 1229–1235. [Google Scholar] [CrossRef]
- Gutierrez-Uzquiza, A.; Arechederra, M.; Molina, I.; Banos, R.; Maia, V.; Benito, M.; Guerrero, C.; Porras, A. C3G down-regulates p38 MAPK activity in response to stress by Rap-1 independent mechanisms: Involvement in cell death. Cell Signal. 2010, 22, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Rivero, S.; Baquero, C.; Hernandez-Cano, L.; Roldan-Etcheverry, J.J.; Gutierrez-Herrero, S.; Fernandez-Infante, C.; Martin-Granado, V.; Anguita, E.; de Pereda, J.M.; Porras, A.; et al. C3G, through its GEF activity, induces megakaryocytic differentiation and proplatelet formation. Cell Commun. Signal. 2018, 16, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannekoek, W.J.; Kooistra, M.R.; Zwartkruis, F.J.; Bos, J.L. Cell-cell junction formation: The role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim. Biophys. Acta 2009, 1788, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooistra, M.R.; Dube, N.; Bos, J.L. Rap1: A key regulator in cell-cell junction formation. J. Cell Sci. 2007, 120, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Encabo, S.; Santos, E.; Guerrero, C. C3G mediated suppression of malignant transformation involves activation of PP2A phosphatases at the subcortical actin cytoskeleton. Exp. Cell Res. 2007, 313, 3881–3891. [Google Scholar] [CrossRef] [Green Version]
- Okino, K.; Nagai, H.; Nakayama, H.; Doi, D.; Yoneyama, K.; Konishi, H.; Takeshita, T. Inactivation of Crk SH3 domain-binding guanine nucleotide-releasing factor (C3G) in cervical squamous cell carcinoma. Int. J. Gynecol. Cancer 2006, 16, 763–771. [Google Scholar] [CrossRef]
- Priego, N.; Arechederra, M.; Sequera, C.; Bragado, P.; Vazquez-Carballo, A.; Gutierrez-Uzquiza, A.; Martin-Granado, V.; Ventura, J.J.; Kazanietz, M.G.; Guerrero, C.; et al. C3G knock-down enhances migration and invasion by increasing Rap1-mediated p38alpha activation, while it impairs tumor growth through p38alpha-independent mechanisms. Oncotarget 2016, 7, 45060–45078. [Google Scholar] [CrossRef] [Green Version]
- Dayma, K.; Radha, V. Cytoskeletal remodeling by C3G to induce neurite-like extensions and inhibit motility in highly invasive breast carcinoma cells. Biochim. Biophys. Acta 2011, 1813, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Hirata, T.; Nagai, H.; Koizumi, K.; Okino, K.; Harada, A.; Onda, M.; Nagahata, T.; Mikami, I.; Hirai, K.; Haraguchi, S.; et al. Amplification, up-regulation and over-expression of C3G (CRK SH3 domain-binding guanine nucleotide-releasing factor) in non-small cell lung cancers. J. Hum. Genet. 2004, 49, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Berzal, J.; Castellano, E.; Martin-Encabo, S.; Gutierrez-Cianca, N.; Hernandez, J.M.; Santos, E.; Guerrero, C. Characterization of p87C3G, a novel, truncated C3G isoform that is overexpressed in chronic myeloid leukemia and interacts with Bcr-Abl. Exp. Cell Res. 2006, 312, 938–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequera, C.; Manzano, S.; Guerrero, C.; Porras, A. How Rap and its GEFs control liver physiology and cancer development. C3G alterations in human hepatocarcinoma. Hepat. Oncol. 2018, 5, HEP05. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Richelme, S.; Avazeri, E.; Audebert, S.; Helmbacher, F.; Dono, R.; Maina, F. Tissue-Specific Gain of RTK Signalling Uncovers Selective Cell Vulnerability during Embryogenesis. PLoS Genet. 2015, 11, e1005533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Arechederra, M.; Richelme, S.; Daian, F.; Novello, C.; Calderaro, J.; Di Tommaso, L.; Morcrette, G.; Rebouissou, S.; Donadon, M.; et al. A phosphokinome-based screen uncovers new drug synergies for cancer driven by liver-specific gain of nononcogenic receptor tyrosine kinases. Hepatology 2017, 66, 1644–1661. [Google Scholar] [CrossRef] [Green Version]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Fan, Y.; Bazai, S.K.; Daian, F.; Arechederra, M.; Richelme, S.; Temiz, N.A.; Yim, A.; Habermann, B.H.; Dono, R.; Largaespada, D.A.; et al. Evaluating the landscape of gene cooperativity with receptor tyrosine kinases in liver tumorigenesis using transposon-mediated mutagenesis. J. Hepatol. 2019, 70, 470–482. [Google Scholar] [CrossRef]
- Cassol, F.; Portal, L.; Richelme, S.; Dupont, M.; Boursier, Y.; Arechederra, M.; Auphan-Anezin, N.; Chasson, L.; Laprie, C.; Fernandez, S.; et al. Tracking dynamics of spontaneous tumors in mice using photon-counting computed tomography. iScience 2019, 21, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Celia, S.; Almudena, P. Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid, Spain. Personal observation, 2020. [Google Scholar]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, J.W.; Gomez, E.W. Biomechanics of TGFbeta-induced epithelial-mesenchymal transition: Implications for fibrosis and cancer. Clin. Transl. Med. 2014, 3, 23. [Google Scholar] [CrossRef]
- Bertran, E.; Crosas-Molist, E.; Sancho, P.; Caja, L.; Lopez-Luque, J.; Navarro, E.; Egea, G.; Lastra, R.; Serrano, T.; Ramos, E.; et al. Overactivation of the TGF-beta pathway confers a mesenchymal-like phenotype and CXCR4-dependent migratory properties to liver tumor cells. Hepatology 2013, 58, 2032–2044. [Google Scholar] [CrossRef]
- Sakkab, D.; Lewitzky, M.; Posern, G.; Schaeper, U.; Sachs, M.; Birchmeier, W.; Feller, S.M. Signaling of hepatocyte growth factor/scatter factor (HGF) to the small GTPase Rap1 via the large docking protein Gab1 and the adapter protein CRKL. J. Biol. Chem. 2000, 275, 10772–10778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamorte, L.; Royal, I.; Naujokas, M.; Park, M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol. Biol. Cell 2002, 13, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Tsuda, M.; Makino, Y.; Konstantinou, T.; Nishihara, H.; Majima, T.; Minami, A.; Feller, S.M.; Tanaka, S. Crk adaptor protein-induced phosphorylation of Gab1 on tyrosine 307 via Src is important for organization of focal adhesions and enhanced cell migration. Cell Res. 2009, 19, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteucci, E.; Ridolfi, E.; Desiderio, M.A. Hepatocyte growth factor differently influences Met-E-cadherin phosphorylation and downstream signaling pathway in two models of breast cells. Cell. Mol. Life Sci. 2006, 63, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Kamei, T.; Matozaki, T.; Sakisaka, T.; Kodama, A.; Yokoyama, S.; Peng, Y.F.; Nakano, K.; Takaishi, K.; Takai, Y. Coendocytosis of cadherin and c-Met coupled to disruption of cell-cell adhesion in MDCK cells-Regulation by Rho, Rac and Rab small G proteins. Oncogene 1999, 18, 6776–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, C.; Serpente, N.; Cogram, P.; Hosking, C.R.; Bialucha, C.U.; Feller, S.M.; Braga, V.M.; Birchmeier, W.; Fujita, Y. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol. Cell. Biol. 2004, 24, 6690–6700. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.H.; Wu, T.T.; Huang, C.Y.; Hsieh, Y.S.; Hwang, J.M.; Liu, J.Y. P38 mitogen-activated protein kinase pathway is involved in protein kinase Calpha-regulated invasion in human hepatocellular carcinoma cells. Cancer Res. 2007, 67, 4320–4327. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, S.; Wang, Z.; Wang, F.; Cao, X.; Wu, Q.; Zhao, C.; Ma, H.; Ye, F.; Wang, H.; et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J. Exp. Clin. Cancer Res. 2018, 37, 128. [Google Scholar] [CrossRef]
- Luk, S.T.; Ng, K.Y.; Zhou, L.; Tong, M.; Wong, T.L.; Yu, H.; Lo, C.M.; Man, K.; Guan, X.Y.; Lee, T.K.; et al. Deficiency in embryonic stem cell marker reduced expression 1 activates mitogen-activated protein kinase kinase 6-dependent p38 mitogen-activated protein kinase signaling to drive hepatocarcinogenesis. Hepatology 2019. [Google Scholar] [CrossRef]
- Dayma, K.; Ramadhas, A.; Sasikumar, K.; Radha, V. Reciprocal Negative Regulation between the Guanine Nucleotide Exchange Factor C3G and beta-Catenin. Genes Cancer 2012, 3, 564–577. [Google Scholar] [CrossRef]
- Lin, Y.; Mettling, C.; Chou, C. Rap1-suppressed tumorigenesis is concomitant with the interference in ras effector signaling. FEBS Lett. 2000, 467, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Ding, S.; Chen, K.; Chen, J.; Wang, S.; Zou, C.; Zhang, J.; Cao, Y.; Huang, A.; Tang, H. Functional analysis of miR-101-3p and Rap1b involved in hepatitis B virus-related hepatocellular carcinoma pathogenesis. Biochem. Cell Biol. 2014, 92, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Duan, H.B.; Yang, Y.S. Knockdown of rap2b inhibits the proliferation and invasion in hepatocellular carcinoma cells. Oncol. Res. 2017, 25, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Peng, H.; Chen, J.; Liu, Y.; Yan, S.; Yu, G.; Chen, Q.; Tang, H.; Liu, S. Rap1b enhances the invasion and migration of hepatocellular carcinoma cells by up-regulating Twist 1. Exp. Cell Res. 2018, 367, 56–64. [Google Scholar] [CrossRef]
- Maia, V.; Ortiz-Rivero, S.; Sanz, M.; Gutierrez-Berzal, J.; Alvarez-Fernandez, I.; Gutierrez-Herrero, S.; de Pereda, J.M.; Porras, A.; Guerrero, C. C3G forms complexes with Bcr-Abl and p38alpha MAPK at the focal adhesions in chronic myeloid leukemia cells: Implication in the regulation of leukemic cell adhesion. Cell Commun. Signal. 2013, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Lamballe, F.; Toscano, S.; Conti, F.; Arechederra, M.; Baeza, N.; Figarella-Branger, D.; Helmbacher, F.; Maina, F. Coordination of signalling networks and tumorigenic properties by ABL in glioblastoma cells. Oncotarget 2016, 7, 74747–74767. [Google Scholar] [CrossRef] [Green Version]
- Asuri, S.; Yan, J.; Paranavitana, N.C.; Quilliam, L.A. E-cadherin dis-engagement activates the Rap1 GTPase. J. Cell Biochem. 2008, 105, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Ma, Y.; Zou, Y.; Yin, A.; Li, W.; Dong, D. HCMDB: The human cancer metastasis database. Nucleic Acids Res. 2018, 46, D950–D955. [Google Scholar] [CrossRef] [Green Version]
- Nebreda, A.R.; Gannon, J.V.; Hunt, T. Newly synthesized protein(s) must associate with p34cdc2 to activate MAP kinase and MPF during progesterone-induced maturation of Xenopus oocytes. EMBO J. 1995, 14, 5597–5607. [Google Scholar] [CrossRef]
- Zuluaga, S.; Gutierrez-Uzquiza, A.; Bragado, P.; Alvarez-Barrientos, A.; Benito, M.; Nebreda, A.R.; Porras, A. P38alpha MAPK can positively or negatively regulate Rac-1 activity depending on the presence of serum. FEBS Lett. 2007, 581, 3819–3825. [Google Scholar] [CrossRef] [Green Version]
- Gebäck, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Al Mahdi, N.; Fazel Najafabadi, M.; Pilarczyck, M.; Kouril, M.; Medvedovic, M. GREIN: An Interactive Web Platform for Re-analyzing GEO RNA-seq Data. Sci. Rep. 2019, 9, 7580. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sequera, C.; Bragado, P.; Manzano, S.; Arechederra, M.; Richelme, S.; Gutiérrez-Uzquiza, A.; Sánchez, A.; Maina, F.; Guerrero, C.; Porras, A. C3G Is Upregulated in Hepatocarcinoma, Contributing to Tumor Growth and Progression and to HGF/MET Pathway Activation. Cancers 2020, 12, 2282. https://doi.org/10.3390/cancers12082282
Sequera C, Bragado P, Manzano S, Arechederra M, Richelme S, Gutiérrez-Uzquiza A, Sánchez A, Maina F, Guerrero C, Porras A. C3G Is Upregulated in Hepatocarcinoma, Contributing to Tumor Growth and Progression and to HGF/MET Pathway Activation. Cancers. 2020; 12(8):2282. https://doi.org/10.3390/cancers12082282
Chicago/Turabian StyleSequera, Celia, Paloma Bragado, Sara Manzano, Maria Arechederra, Sylvie Richelme, Alvaro Gutiérrez-Uzquiza, Aránzazu Sánchez, Flavio Maina, Carmen Guerrero, and Almudena Porras. 2020. "C3G Is Upregulated in Hepatocarcinoma, Contributing to Tumor Growth and Progression and to HGF/MET Pathway Activation" Cancers 12, no. 8: 2282. https://doi.org/10.3390/cancers12082282
APA StyleSequera, C., Bragado, P., Manzano, S., Arechederra, M., Richelme, S., Gutiérrez-Uzquiza, A., Sánchez, A., Maina, F., Guerrero, C., & Porras, A. (2020). C3G Is Upregulated in Hepatocarcinoma, Contributing to Tumor Growth and Progression and to HGF/MET Pathway Activation. Cancers, 12(8), 2282. https://doi.org/10.3390/cancers12082282