Retinoblastoma: Etiology, Modeling, and Treatment


Abstract
:1. Introduction
2. Molecular and Cellular Basis of Retinoblastoma
2.1. Genomic Landscape
2.2. Gene Expression Profile of Tumor Tissue from Enucleated Eyes
2.3. Correlation of Genomic and Transcriptomic Profiles with Clinicopathological Characteristics
2.4. Retinoblastoma Cell-of-Origin
2.4.1. Cone Cells Proliferate in Response to Retinoblastoma Protein (pRb) Loss, While Cone-Enriched Genes are Prominently Expressed in Retinoblastoma
2.4.2. Cone-Specific Signal Circuitry Promotes Retinoblastoma Genesis
3. Disease Modeling
3.1. Genetically Engineered Mouse Models (GEMMs)
3.2. Xenografts and Organoids
3.3. Advantages and Limitations
4. Treatments
4.1. Classification and Staging Systems
4.2. High-Risk Histopathological Features
4.3. Current Treatments
4.4. Clinicopathological Correlations
4.5. Future Treatments
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojanaporn, D.; Boontawon, T.; Chareonsirisuthigul, T.; Thanapanpanich, O.; Attaseth, T.; Saengwimol, D.; Anurathapan, U.; Sujirakul, T.; Kaewkhaw, R.; Hongeng, S. Spectrum of germline RB1 mutations and clinical manifestations in retinoblastoma patients from Thailand. Mol. Vis. 2018, 24, 778–788. [Google Scholar] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampieri, K.; Amenduni, M.; Papa, F.T.; Katzaki, E.; Mencarelli, M.A.; Marozza, A.; Epistolato, M.C.; Toti, P.; Lazzi, S.; Bruttini, M.; et al. Array comparative genomic hybridization in retinoma and retinoblastoma tissues. Cancer Sci. 2009, 100, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef]
- Abramson, D.H.; Frank, C.M.; Susman, M.; Whalen, M.P.; Dunkel, I.J.; Boyd, N.W., 3rd. Presenting signs of retinoblastoma. J. Pediatrics 1998, 132, 505–508. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Berry, J.L.; Kogachi, K.; Aziz, H.A.; McGovern, K.; Zolfaghari, E.; Murphree, A.L.; Jubran, R.; Kim, J.W. Risk of metastasis and orbital recurrence in advanced retinoblastoma eyes treated with systemic chemoreduction versus primary enucleation. Pediatric Blood Cancer 2017, 64, e26270. [Google Scholar] [CrossRef]
- Kaliki, S.; Mittal, P.; Mohan, S.; Chattannavar, G.; Jajapuram, S.D.; Mohamed, A.; Palkonda, V.A.R. Bilateral advanced (Group D or E) intraocular retinoblastoma: Outcomes in 72 Asian Indian patients. Eye 2019, 33, 1297–1304. [Google Scholar] [CrossRef]
- Perez, V.; Sampor, C.; Rey, G.; Parareda-Salles, A.; Kopp, K.; Dabezies, A.P.; Dufort, G.; Zelter, M.; Lopez, J.P.; Urbieta, M.; et al. Treatment of nonmetastatic unilateral retinoblastoma in children. JAMA Ophthalmol. 2018, 136, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Chevez-Barrios, P.; Eagle, R.C.; Krailo, M.; Piao, J.; Albert, D.M.; Gao, Y.; Vemuganti, G.; Ali, M.J.; Khetan, V.; Honavar, S.G.; et al. Study of unilateral retinoblastoma with and without histopathologic high-risk features and the role of adjuvant chemotherapy: A Children’s Oncology Group study. J. Clin. Oncol. 2019, 37, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Brodie, S.E.; Marr, B.; Zabor, E.C.; Mondesire-Crump, I.; Abramson, D.H. Efficacy and toxicity of intravitreous chemotherapy for retinoblastoma: Four-year experience. Ophthalmology 2017, 124, 488–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, J.H.; Schaiquevich, P.; Buitrago, E.; Del Sole, M.J.; Zapata, G.; Croxatto, J.O.; Marr, B.P.; Brodie, S.E.; Berra, A.; Chantada, G.L.; et al. Local and systemic toxicity of intravitreal melphalan for vitreous seeding in retinoblastoma: A preclinical and clinical study. Ophthalmology 2014, 121, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Saakyan, S.; Myakoshina, E.; Ismailova, D. Retinopathy in young retinoblastoma patients receiving a chemotherapy treatment: Clinical trials and morphometric analysis. Ophthalmic Genet. 2019, 40, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.E.; D’Silva, C.N.; Dimaras, H.; Dzneladze, I.; Chan, H.; Gallie, B.L. Clinical and genetic associations for carboplatin-related ototoxicity in children treated for retinoblastoma: A retrospective noncomparative single-institute experience. Pediatric Blood Cancer 2018, 65, e26931. [Google Scholar] [CrossRef]
- Shields, C.L.; Lally, S.E.; Leahey, A.M.; Jabbour, P.M.; Caywood, E.H.; Schwendeman, R.; Shields, J.A. Targeted retinoblastoma management: When to use intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Curr. Opin. Ophthalmol. 2014, 25, 374–385. [Google Scholar] [CrossRef]
- Singh, H.P.; Wang, S.; Stachelek, K.; Lee, S.; Reid, M.W.; Thornton, M.E.; Craft, C.M.; Grubbs, B.H.; Cobrinik, D. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc. Natl. Acad. Sci. USA 2018, 115, E9391–E9400. [Google Scholar] [CrossRef] [Green Version]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivela, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef] [Green Version]
- Coschi, C.H.; Ishak, C.A.; Gallo, D.; Marshall, A.; Talluri, S.; Wang, J.; Cecchini, M.J.; Martens, A.L.; Percy, V.; Welch, I.; et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 2014, 4, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.L.; Benes, C.; Dyson, N.J. Whole chromosome instability resulting from the synergistic effects of pRB and p53 inactivation. Oncogene 2014, 33, 2487–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Harn, T.; Foijer, F.; van Vugt, M.; Banerjee, R.; Yang, F.; Oostra, A.; Joenje, H.; te Riele, H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010, 24, 1377–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooi, I.E.; Mol, B.M.; Massink, M.P.; Ameziane, N.; Meijers-Heijboer, H.; Dommering, C.J.; van Mil, S.E.; de Vries, Y.; van der Hout, A.H.; Kaspers, G.J.; et al. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci. Rep. 2016, 6, 25264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, J.; Nagahawatte, P.; Finkelstein, D.; Richards-Yutz, J.; Valentine, M.; Ma, J.; Mullighan, C.; Song, G.; Chen, X.; Wilson, M.; et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 2014, 5, 438–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, L.; Xu, X.L.; Abramson, D.H.; Jhanwar, S.C. Genomic instability and proliferation/survival pathways in RB1-deficient malignancies. Adv. Biol. Regul. 2017, 64, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Singh, H.P.; Wang, L.; Qi, D.L.; Poulos, B.K.; Abramson, D.H.; Jhanwar, S.C.; Cobrinik, D. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 2014, 514, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.N.; Gallie, B.L.; Squire, J.A. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet. Cytogenet. 2001, 129, 57–63. [Google Scholar] [CrossRef]
- Herzog, S.; Lohmann, D.R.; Buiting, K.; Schuler, A.; Horsthemke, B.; Rehder, H.; Rieder, H. Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization. Hum. Genet. 2001, 108, 98–104. [Google Scholar] [CrossRef]
- Lillington, D.M.; Kingston, J.E.; Coen, P.G.; Price, E.; Hungerford, J.; Domizio, P.; Young, B.D.; Onadim, Z. Comparative genomic hybridization of 49 primary retinoblastoma tumors identifies chromosomal regions associated with histopathology, progression, and patient outcome. Genes Chromosomes Cancer 2003, 36, 121–128. [Google Scholar] [CrossRef]
- Mairal, A.; Pinglier, E.; Gilbert, E.; Peter, M.; Validire, P.; Desjardins, L.; Doz, F.; Aurias, A.; Couturier, J. Detection of chromosome imbalances in retinoblastoma by parallel karyotype and CGH analyses. Gene Chromosome Cancer 2000, 28, 370–379. [Google Scholar] [CrossRef]
- Mol, B.M.; Massink, M.P.; van der Hout, A.H.; Dommering, C.J.; Zaman, J.M.; Bosscha, M.I.; Kors, W.A.; Meijers-Heijboer, H.E.; Kaspers, G.J.; Riele, H.; et al. High resolution SNP array profiling identifies variability in retinoblastoma genome stability. Genes Chromosomes Cancer 2014, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, B.; Gratias, S.; Toedt, G.; Mendrzyk, F.; Stange, D.E.; Radlwimmer, B.; Lohmann, D.R.; Lichter, P. Detection of chromosomal imbalances in retinoblastoma by matrix-based comparative genomic hybridization. Genes Chromosomes Cancer 2005, 43, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Kooi, I.E.; Mol, B.M.; Massink, M.P.; de Jong, M.C.; de Graaf, P.; van der Valk, P.; Meijers-Heijboer, H.; Kaspers, G.J.; Moll, A.C.; te Riele, H.; et al. A meta-analysis of retinoblastoma copy numbers refines the list of possible driver genes involved in tumor progression. PLoS ONE 2016, 11, e0153323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratias, S.; Rieder, H.; Ullmann, R.; Klein-Hitpass, L.; Schneider, S.; Boloni, R.; Kappler, M.; Lohmann, D.R. Allelic loss in a minimal region on chromosome 16q24 is associated with vitreous seeding of retinoblastoma. Cancer Res. 2007, 67, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Gustmann, S.; Klein-Hitpass, L.; Stephan, H.; Weber, S.; Bornfeld, N.; Kaulisch, M.; Lohmann, D.R.; Dünker, N. Loss at chromosome arm 16q in retinoblastoma: Confirmation of the association with diffuse vitreous seeding and refinement of the recurrently deleted region. Genes Chromosomes Cancer 2011, 50, 327–337. [Google Scholar] [CrossRef]
- Marchong, M.N.; Chen, D.; Corson, T.W.; Lee, C.; Harmandayan, M.; Bowles, E.; Chen, N.; Gallie, B.L. Minimal 16q genomic loss implicates cadherin-11 in retinoblastoma. Mol. Cancer Res. 2004, 2, 495–503. [Google Scholar]
- Ewens, K.G.; Bhatti, T.R.; Moran, K.A.; Richards-Yutz, J.; Shields, C.L.; Eagle, R.C.; Ganguly, A. Phosphorylation of pRb: Mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med. 2017, 6, 619–630. [Google Scholar] [CrossRef]
- Polski, A.; Xu, L.; Prabakar, R.K.; Gai, X.; Kim, J.W.; Shah, R.; Jubran, R.; Kuhn, P.; Cobrinik, D.; Hicks, J.; et al. Variability in retinoblastoma genome stability is driven by age and not heritability. Genes Chromosom. Cancer 2020. [Google Scholar] [CrossRef]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Theriault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; Van Ziffle, J.; Onodera, C.; et al. Next-generation sequencing of retinoblastoma identifies pathogenic alterations beyond RB1 inactivation that correlate with aggressive histopathologic features. Ophthalmology 2019. [Google Scholar] [CrossRef]
- Castéra, L.; Sabbagh, A.; Dehainault, C.; Michaux, D.; Mansuet-Lupo, A.; Patillon, B.; Lamar, E.; Aerts, I.; Lumbroso-Le Rouic, L.; Couturier, J.; et al. MDM2 as a modifier gene in retinoblastoma. J. Natl. Cancer Inst. 2010, 102, 1805–1808. [Google Scholar] [CrossRef] [PubMed]
- Laurie, N.A.; Donovan, S.L.; Shih, C.S.; Zhang, J.K.; Mills, N.; Fuller, C.; Teunisse, A.; Lam, S.; Ramos, Y.; Mohan, A.; et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006, 444, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.E.; Mendoza, P.; Hudson, W.H.; Ziesel, A.; Hubbard, G.B., 3rd; Wells, J.; Dwivedi, B.; Kowalski, J.; Seby, S.; Patel, V.; et al. Distinct gene expression profiles define anaplastic grade in retinoblastoma. Am. J. Pathol. 2018, 188, 2328–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapatai, G.; Brundler, M.A.; Jenkinson, H.; Kearns, P.; Parulekar, M.; Peet, A.C.; McConville, C.M. Gene expression profiling identifies different sub-types of retinoblastoma. Br. J. Cancer 2013, 109, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooi, I.E.; Mol, B.M.; Moll, A.C.; van der Valk, P.; de Jong, M.C.; de Graaf, P.; van Mil, S.E.; Schouten-van Meeteren, A.Y.; Meijers-Heijboer, H.; Kaspers, G.L.; et al. Loss of photoreceptorness and gain of genomic alterations in retinoblastoma reveal tumor progression. EBioMedicine 2015, 2, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.L.; Fang, Y.; Lee, T.C.; Forrest, D.; Gregory-Evans, C.; Almeida, D.; Liu, A.; Jhanwar, S.C.; Abramson, D.H.; Cobrinik, D. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 2009, 137, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, A.; Shields, C.L. Differential gene expression profile of retinoblastoma compared to normal retina. Mol. Vis. 2010, 16, 1292–1303. [Google Scholar]
- Saengwimol, D.; Rojanaporn, D.; Chaitankar, V.; Chittavanich, P.; Aroonroch, R.; Boontawon, T.; Thammachote, W.; Jinawath, N.; Hongeng, S.; Kaewkhaw, R. A three-dimensional organoid model recapitulates tumorigenic aspects and drug responses of advanced human retinoblastoma. Sci. Rep. 2018, 8, 15664. [Google Scholar] [CrossRef]
- Chakraborty, S.; Khare, S.; Dorairaj, S.K.; Prabhakaran, V.C.; Prakash, D.R.; Kumar, A. Identification of genes associated with tumorigenesis of retinoblastoma by microarray analysis. Genomics 2007, 90, 344–353. [Google Scholar] [CrossRef]
- McEvoy, J.; Flores-Otero, J.; Zhang, J.; Nemeth, K.; Brennan, R.; Bradley, C.; Krafcik, F.; Rodriguez-Galindo, C.; Wilson, M.; Xiong, S.; et al. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell 2011, 20, 260–275. [Google Scholar] [CrossRef] [Green Version]
- Eagle, R.C., Jr. High-risk features and tumor differentiation in retinoblastoma: A retrospective histopathologic study. Arch. Pathol. Lab. Med. 2009, 133, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Chawla, B.; Sharma, S.; Sen, S.; Azad, R.; Bajaj, M.S.; Kashyap, S.; Pushker, N.; Ghose, S. Correlation between clinical features, magnetic resonance imaging, and histopathologic findings in retinoblastoma: A prospective study. Ophthalmology 2012, 119, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.W.; de Jong, M.C.; Kooi, I.E.; Sirin, S.; Goricke, S.; Brisse, H.J.; Maeder, P.; Galluzzi, P.; van der Valk, P.; Cloos, J.; et al. MR imaging features of retinoblastoma: Association with gene expression profiles. Radiology 2018, 288, 506–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Turaka, K.; Shields, C.L. Retinocytoma shows lack of response to chemoreduction. J. Pediatric Ophthalmol. Strabismus 2010, 47, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Demirci, H.; Eagle, R.C., Jr.; Shields, C.L.; Shields, J.A. Histopathologic findings in eyes with retinoblastoma treated only with chemoreduction. Arch. Ophthalmol. 2003, 121, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.D.; Shields, C.L.; Shields, J.A. Lack of response to chemoreduction in presumed well differentiated retinoblastoma. J. Pediatric Ophthalmol. Strabismus 2002, 39, 107–109. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, U.M.; O’Brien, J.M. Understanding pRb: Toward the necessary development of targeted treatments for retinoblastoma. J. Clin. Investig. 2012, 122, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.C.; Almeida, D.; Claros, N.; Abramson, D.H.; Cobrinik, D. Cell cycle-specific and cell type-specific expression of Rb in the developing human retina. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5590–5598. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gray, J.; Wu, L.; Leone, G.; Rowan, S.; Cepko, C.L.; Zhu, X.; Craft, C.M.; Dyer, M.A. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nat. Genet. 2004, 36, 351–360. [Google Scholar] [CrossRef]
- Furukawa, T.; Morrow, E.M.; Li, T.; Davis, F.C.; Cepko, C.L. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet. 1999, 23, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Furukawa, A.; Koike, C.; Tano, Y.; Aizawa, S.; Matsuo, I.; Furukawa, T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 2003, 6, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Glubrecht, D.D.; Kim, J.H.; Russell, L.; Bamforth, J.S.; Godbout, R. Differential CRX and OTX2 expression in human retina and retinoblastoma. J. Neurochem. 2009, 111, 250–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.R.; Hendrickson, A.; McGuire, C.R.; Reh, T.A. Retinoid X receptor gamma is necessary to establish the S-opsin gradient in cone photoreceptors of the developing mouse retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2897–2904. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.; Hurley, J.B.; Dierks, B.; Srinivas, M.; Saltó, C.; Vennström, B.; Reh, T.A.; Forrest, D. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 2001, 27, 94–98. [Google Scholar] [CrossRef]
- Ng, L.; Lu, A.L.; Swaroop, A.; Sharlin, D.S.; Swaroop, A.; Forrest, D. Two transcription factors can direct three photoreceptor outcomes from rod precursor cells in mouse retinal development. J. Neurosci. 2011, 31, 11118–11125. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Pajovic, S.; Gallie, B.L. Expression of p14ARF, MDM2, and MDM4 in human retinoblastoma. Biochem. Biophys. Res. Commun. 2008, 375, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Qi, D.L.; Singh, H.P.; Zou, Y.; Shen, B.; Cobrinik, D. A novel thyroid hormone receptor isoform, TRbeta2-46, promotes SKP2 expression and retinoblastoma cell proliferation. J. Biol. Chem. 2019, 294, 2961–2969. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.L.; Cobrinik, D. MDM2 but not MDM4 promotes retinoblastoma cell proliferation through p53-independent regulation of MYCN translation. Oncogene 2017, 36, 1760–1769. [Google Scholar] [CrossRef] [Green Version]
- To, K.H.; Pajovic, S.; Gallie, B.L.; Theriault, B.L. Regulation of p14ARF expression by miR-24: A potential mechanism compromising the p53 response during retinoblastoma development. BMC Cancer 2012, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.L.; Li, Z.; Liu, A.; Fan, X.; Hu, D.N.; Qi, D.L.; Chitty, D.W.; Jia, R.; Qui, J.; Wang, J.Q.; et al. SKP2 activation by thyroid hormone receptor beta2 bypasses Rb-dependent proliferation in Rb-deficient cells. Cancer Res. 2017, 77, 6838–6850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Taya, Y.; Nakagama, H. Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS Lett. 2009, 583, 2710–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, D.; Lukas, J.; Reed, S.I. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein–ubiquitin ligase SCFSkp2. Genes Dev. 2002, 16, 2946–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.L.; Bauzon, F.; Fu, H.; Cui, J.H.; Zhao, H.L.; Nakayama, K.; Nakayama, K.I.; Zhu, L. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat. Commun. 2014, 5, 3463. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.B.; Bauzon, F.; Ji, P.; Xu, X.L.; Sun, D.Q.; Locker, J.; Sellers, R.S.; Nakayama, K.; Nakayama, K.I.; Cobrinik, D.; et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1(+/−) mice. Nat. Genet. 2010, 42, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Iglesias, O.; Garcia-Silva, S.; Tenbaum, S.P.; Regadera, J.; Larcher, F.; Paramio, J.M.; Vennström, B.; Aranda, A. Thyroid hormone receptor β1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009, 69, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.V.; Shimizu, T.; Ishizaki, K.; Kaneko, A.; Yandell, D.W.; Toguchida, J.; Sasaki, M.S. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996, 106, 75–82. [Google Scholar] [CrossRef]
- Ruiz-Perez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN protein in health and disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Lee, W.H.; Murphree, A.L.; Benedict, W.F. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, D.; Conkrite, K.; Tam, M.; Mukai, S.; Mu, D.; Jacks, T. Murine bilateral retinoblastoma exhibiting rapid-onset, metastatic progression and N-myc gene amplification. EMBO J. 2007, 26, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Jia, D.; Bates, B.; Basom, R.; Eberhart, C.G.; MacPherson, D. A mouse model of MYCN-driven retinoblastoma reveals MYCN-independent tumor reemergence. J. Clin. Investig. 2017, 127, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.L.; Schweers, B.; Martins, R.; Johnson, D.; Dyer, M.A. Compensation by tumor suppressor genes during retinal development in mice and humans. BMC Biol. 2006, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, D.; Sage, J.; Kim, T.; Ho, D.; McLaughlin, M.E.; Jacks, T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004, 18, 1681–1694. [Google Scholar] [CrossRef] [Green Version]
- Dannenberg, J.H.; Schuijff, L.; Dekker, M.; van der Valk, M.; te Riele, H. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004, 18, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Ajioka, I.; Martins, R.A.; Bayazitov, I.T.; Donovan, S.; Johnson, D.A.; Frase, S.; Cicero, S.A.; Boyd, K.; Zakharenko, S.S.; Dyer, M.A. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell 2007, 131, 378–390. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, D.; Dyer, M.A. Retinoblastoma: From the two-hit hypothesis to targeted chemotherapy. Cancer Res. 2007, 67, 7547–7550. [Google Scholar] [CrossRef] [Green Version]
- Kooi, I.E.; van Mil, S.E.; MacPherson, D.; Mol, B.M.; Moll, A.C.; Meijers-Heijboer, H.; Kaspers, G.J.; Cloos, J.; Te Riele, H.; Dorsman, J.C. Genomic landscape of retinoblastoma in Rb(−/−) p130(−/−) mice resembles human retinoblastoma. Genes Chromosomes Cancer 2017, 56, 231–242. [Google Scholar] [CrossRef]
- Zhang, J.; Schweers, B.; Dyer, M.A. The first knockout mouse model of retinoblastoma. Cell Cycle 2004, 3, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Vooijs, M.; te Riele, H.; van der Valk, M.; Berns, A. Tumor formation in mice with somatic inactivation of the retinoblastoma gene in interphotoreceptor retinol binding protein-expressing cells. Oncogene 2002, 21, 4635–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benavente, C.A.; McEvoy, J.D.; Finkelstein, D.; Wei, L.; Kang, G.; Wang, Y.D.; Neale, G.; Ragsdale, S.; Valentine, V.; Bahrami, A.; et al. Cross-species genomic and epigenomic landscape of retinoblastoma. Oncotarget 2013, 4, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Lu, H.; Nomura, A.; Hanse, E.A.; Forster, C.L.; Parker, J.B.; Linden, M.A.; Karasch, C.; Hallstrom, T.C. Co-deleting Pten with Rb in retinal progenitor cells in mice results in fully penetrant bilateral retinoblastomas. Mol. Cancer 2015, 14, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, I.; Leuraud, P.; Blais, J.; Pouliquen, A.L.; Maillard, P.; Houdayer, C.; Couturier, J.; Sastre-Garau, X.; Grierson, D.; Doz, F.; et al. In vivo efficacy of photodynamic therapy in three new xenograft models of human retinoblastoma. Photodiagn. Photodyn. Ther. 2010, 7, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Cassoux, N.; Thuleau, A.; Assayag, F.; Aerts, I.; Decaudin, D. Establishment of an Orthotopic Xenograft Mice Model of Retinoblastoma Suitable for Preclinical Testing. Ocul. Oncol. Pathol. 2015, 1, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Lemaitre, S.; Poyer, F.; Marco, S.; Fréneaux, P.; Doz, F.; Aerts, I.; Desjardins, L.; Cassoux, N.; Thomas, C.D. Looking for the most suitable orthotopic retinoblastoma mouse model in order to characterize the tumoral development. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3055–3064. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Pasto, G.; Olaciregui, N.G.; Vila-Ubach, M.; Paco, S.; Monterrubio, C.; Rodriguez, E.; Winter, U.; Batalla-Vilacis, M.; Catala, J.; Salvador, H.; et al. Preclinical platform of retinoblastoma xenografts recapitulating human disease and molecular markers of dissemination. Cancer Lett. 2016, 380, 10–19. [Google Scholar] [CrossRef]
- Cui, Z.; Guo, Y.; Zhou, Y.; Mao, S.; Yan, X.; Zeng, Y.; Ding, C.; Chan, H.f.; Tang, S.; Tang, L.; et al. Transcriptomic analysis of the developmental similarities and differences between the native retina and retinal organoids. Investig. Ophthalmol. Vis. Sci. 2020, 61, 6. [Google Scholar] [CrossRef] [Green Version]
- Kaewkhaw, R.; Kaya, K.D.; Brooks, M.; Homma, K.; Zou, J.; Chaitankar, V.; Rao, M.; Swaroop, A. Transcriptome dynamics of developing photoreceptors in three-dimensional retina cultures recapitulates temporal sequence of human cone and rod differentiation revealing cell surface markers and gene networks. Stem Cells 2015, 33, 3504–3518. [Google Scholar] [CrossRef]
- Kaewkhaw, R.; Swaroop, M.; Homma, K.; Nakamura, J.; Brooks, M.; Kaya, K.D.; Chaitankar, V.; Michael, S.; Tawa, G.; Zou, J.Z.; et al. Treatment paradigms for retinal and macular diseases using 3-D retina cultures derived from human reporter pluripotent stem cell lines. Investig. Ophthalmol. Vis. Sci. 2016, 57, ORSFl1–ORSFl11. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 116, 10824–10833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-cell transcriptomic comparison of human fetal retina, hPSC-derived retinal organoids, and long-term retinal cultures. Cell Rep. 2020, 30, 1644–1659.e1644. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Khetan, V.; Rishi, P.; Suganeswari, G.; Krishnakumar, S.; Krishnan, U.M.; Parameswaran, S. Generation of a human induced pluripotent stem cell line (VRFi001-A) from orbital adipose tissue of a bilateral retinoblastoma patient with heterozygous RB1 gene deletion. Stem Cell Res. 2018, 29, 42–45. [Google Scholar] [CrossRef]
- Yue, F.M.; Hirashima, K.; Tomotsune, D.; Takizawa-Shirasawa, S.; Yokoyama, T.; Sasaki, K. Reprogramming of retinoblastoma cancer cells into cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 482, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.C.; Liu, L.J.; Ouyang, Q.; Zhao, Y.; Lin, G.; Hu, L.; Li, W. Generation of induced pluripotent stem cells (iPSCs) from a retinoblastoma patient carrying a c.2663G > A mutation in RB1 gene. Stem Cell Res. 2016, 17, 208–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avior, Y.; Lezmi, E.; Yanuka, D.; Benvenisty, N. Modeling developmental and tumorigenic aspects of trilateral retinoblastoma via human embryonic stem cells. Stem Cell Rep. 2017, 8, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Steenpass, L. Generation of two H1 hESC sublines carrying a heterozygous and homozygous knock-out of RB1. Stem Cell Res. 2017, 25, 270–273. [Google Scholar] [CrossRef]
- Reese, A.B.; Ellsworth, R.M. The evaluation and current concept of retinoblastoma therapy. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1963, 67, 164–172. [Google Scholar]
- Murphree, L.A. Intraocular retinoblastoma: The case for a new group classification. Ophthalmol. Clin. North Am. 2005, 18, 41–53. [Google Scholar] [CrossRef]
- Shields, C.L.; Mashayekhi, A.; Au, A.K.; Czyz, C.; Leahey, A.; Meadows, A.T.; Shields, J.A. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 2006, 113, 2276–2280. [Google Scholar] [CrossRef]
- Fabian, I.D.; Reddy, A.; Sagoo, M.S. Classification and staging of retinoblastoma. Commun. Eye Health 2018, 31, 11–13. [Google Scholar]
- Novetsky, D.E.; Abramson, D.H.; Kim, J.W.; Dunkel, I.J. Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet. 2009, 30, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Mallipatna, A.C.; Gallie, B.L.; Chévez-Barrios, P.; Lumbroso-Le Rouic, L.; Chantada, G.; Brisse, H.J.; Doz, F.; Munier, F.L.; Albert, D.M.; Català-Mora, J.; et al. Retinoblastoma. In AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Edge, S.B., Greene, F.L., Byrd, D.R., Brookland, R.K., Washington, M.K., Gershenwald, J.E., Compton, C.C., Hess, K.R., Sullivan, D.C., et al., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 819–831. [Google Scholar]
- Kaliki, S.; Shields, C.L.; Rojanaporn, D.; Al-Dahmash, S.; McLaughlin, J.P.; Shields, J.A.; Eagle, R.C. High-risk retinoblastoma based on international classification of retinoblastoma: Analysis of 519 enucleated eyes. Ophthalmology 2013, 120, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Kaliki, S.; Srinivasan, V.; Gupta, A.; Mishra, D.K.; Naik, M.N. Clinical features predictive of high-risk retinoblastoma in 403 Asian Indian patients: A case-control study. Ophthalmology 2015, 122, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Sastre, X.; Chantada, G.L.; Doz, F.; Wilson, M.W.; de Davila, M.T.; Rodriguez-Galindo, C.; Chintagumpala, M.; Chevez-Barrios, P.; International Retinoblastoma Staging Working Group. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch. Pathol. Lab. Med. 2009, 133, 1199–1202. [Google Scholar] [CrossRef]
- Fletcher, O.; Easton, D.; Anderson, K.; Gilham, C.; Jay, M.; Peto, J. Lifetime risks of common cancers among retinoblastoma survivors. J. Natl. Cancer Inst. 2004, 96, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Kleinerman, R.A.; Tucker, M.A.; Tarone, R.E.; Abramson, D.H.; Seddon, J.M.; Stovall, M.; Li, F.P.; Fraumeni, J.F. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: An extended follow-up. J. Clin. Oncol. 2005, 23, 2272–2279. [Google Scholar] [CrossRef]
- Berry, J.L.; Jubran, R.; Kim, J.W.; Wong, K.; Bababeygy, S.R.; Almarzouki, H.; Lee, T.C.; Murphree, A.L. Long-term outcomes of group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRTsalvage. Pediatric Blood Cancer 2013, 60, 688–693. [Google Scholar] [CrossRef]
- Yamane, T.; Kaneko, A.; Mohri, M. The technique of ophthalmic arterial infusion therapy for patients with intraocular retinoblastoma. Int. J. Clin. Oncol. 2004, 9, 69–73. [Google Scholar] [CrossRef]
- Abramson, D.H.; Dunkel, I.J.; Brodie, S.E.; Kim, J.W.; Gobin, Y.P. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma initial results. Ophthalmology 2008, 115, 1398–1404.e1. [Google Scholar] [CrossRef]
- Abramson, D.H.; Daniels, A.B.; Marr, B.P.; Francis, J.H.; Brodie, S.E.; Dunkel, I.J.; Gobin, Y.P. Intra-arterial chemotherapy (ophthalmic artery chemosurgery) for Group D retinoblastoma. PLoS ONE 2016, 11, e0146582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojanaporn, D.; Chanthanaphak, E.; Boonyaopas, R.; Sujirakul, T.; Hongeng, S.; Ayudhaya, S.S.N. Intra-arterial chemotherapy for retinoblastoma: 8-year experience from a tertiary referral institute in Thailand. Asia Pac. J. Ophthalmol. 2019, 8, 211–217. [Google Scholar] [CrossRef]
- Abramson, D.H.; Marr, B.P.; Dunkel, I.J.; Brodie, S.; Zabor, E.C.; Driscoll, S.J.; Gobin, Y.P. Intra-arterial chemotherapy for retinoblastoma in eyes with vitreous and/or subretinal seeding: 2-year results. Br. J. Ophthalmol. 2012, 96, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Shields, C.L.; Bianciotto, C.G.; Jabbour, P.; Ramasubramanian, A.; Lally, S.E.; Griffin, G.C.; Rosenwasser, R.; Shields, J.A. Intra-arterial chemotherapy for retinoblastoma report no. 1, control of retinal tumors, subretinal seeds, and vitreous seeds. Arch. Ophthalmol. 2011, 129, 1399–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seregard, S.; Kock, E.; af Trampe, E. Intravitreal chemotherapy for recurrent retinoblastoma in an only eye. Br. J. Ophthalmol. 1995, 79, 194–195. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, A.; Suzuki, S. Eye-preservation treatment of retinoblastoma with vitreous seeding. Jpn. J. Clin. Oncol. 2003, 33, 601–607. [Google Scholar] [CrossRef]
- Munier, F.L.; Soliman, S.; Moulin, A.P.; Gaillard, M.C.; Balmer, A.; Beck-Popovic, M. Profiling safety of intravitreal injections for retinoblastoma using an anti-reflux procedure and sterilisation of the needle track. Br. J. Ophthalmol. 2012, 96, 1084–1087. [Google Scholar] [CrossRef]
- Winter, U.; Nicolas, M.; Sgroi, M.; Sampor, C.; Torbidoni, A.; Fandino, A.; Chantada, G.L.; Munier, F.L.; Schaiquevich, P. Assessment of retinoblastoma RNA reflux after intravitreal injection of melphalan. Br. J. Ophthalmol. 2018, 102, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Ghassemi, F.; Shields, C.L.; Ghadimi, H.; Khodabandeh, A.; Roohipoor, R. Combined intravitreal melphalan and topotecan for refractory or recurrent vitreous seeding from retinoblastoma. JAMA Ophthalmol. 2014, 132, 936–941. [Google Scholar] [CrossRef] [Green Version]
- Shields, C.L.; Alset, A.E.; Say, E.A.T.; Caywood, E.; Jabbour, P.; Shields, J.A. Retinoblastoma control with primary intra-arterial chemotherapy: Outcomes before and during the intravitreal chemotherapy era. J. Pediatric Ophthalmol. Strabismus 2016, 53, 275–284. [Google Scholar] [CrossRef]
- Rao, R.; Honavar, S.G.; Sharma, V.; Reddy, V.A.P. Intravitreal topotecan in the management of refractory and recurrent vitreous seeds in retinoblastoma. Br. J. Ophthalmol. 2018, 102, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Smith, B.D. Evaluating the risk of extraocular tumour spread following intravitreal injection therapy for retinoblastoma: A systematic review. Br. J. Ophthalmol. 2013, 97, 1231–1236. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.L.; Shah, S.; Bechtold, M.; Zolfaghari, E.; Jubran, R.; Kim, J. Long-term outcomes of Group D retinoblastoma eyes during the intravitreal melphalan era. Pediatric Blood Cancer 2017, 64, e26696. [Google Scholar] [CrossRef] [PubMed]
- Munier, F.L.; Gaillard, M.C.; Balmer, A.; Soliman, S.; Podilsky, G.; Moulin, A.P.; Beck-Popovic, M. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: From prohibition to conditional indications. Br. J. Ophthalmol. 2012, 96, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Aihara, Y.; Fujiwara, M.; Sano, S.; Kaneko, A. Intravitreal injection of melphalan for intraocular retinoblastoma. Jpn. J. Ophthalmol. 2015, 59, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Yousef, Y.A.; Soliman, S.E.; Astudillo, P.P.P.; Durairaj, P.; Dimaras, H.; Chan, H.S.L.; Heon, E.; Gallie, B.L.; Shaikh, F. Intra-arterial chemotherapy for retinoblastoma: A systematic review. JAMA Ophthalmol. 2016, 134, 584–591. [Google Scholar] [CrossRef]
- Munier, F.L.; Mosimann, P.; Puccinelli, F.; Gaillard, M.C.; Stathopoulos, C.; Houghton, S.; Bergin, C.; Beck-Popovic, M. First-line intra-arterial versus intravenous chemotherapy in unilateral sporadic group D retinoblastoma: Evidence of better visual outcomes, ocular survival and shorter time to success with intra-arterial delivery from retrospective review of 20 years of treatment. Br. J. Ophthalmol. 2017, 101, 1086–1093. [Google Scholar] [CrossRef]
- Chantada, G.; Fandino, A.; Davila, M.T.; Manzitti, J.; Raslawski, E.; Casak, S.; Schvartzman, E. Results of a prospective study for the treatment of retinoblastoma. Cancer 2004, 100, 834–842. [Google Scholar] [CrossRef]
- Khelfaoui, F.; Validire, P.; Auperin, A.; Quintana, E.; Michon, J.; Pacquement, H.; Desjardins, L.; Asselain, B.; Schlienger, P.; Vielh, P.; et al. Histopathologic risk factors in retinoblastoma: A retrospective study of 172 patients treated in a single institution. Cancer 1996, 77, 1206–1213. [Google Scholar] [CrossRef]
- Honavar, S.G.; Singh, A.D.; Shields, C.L.; Meadows, A.T.; Demirci, H.; Cater, J.; Shields, J.A. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch. Ophthalmol. 2002, 120, 923–931. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L.; Shah, S.U.; Eagle, R.C., Jr.; Shields, J.A.; Leahey, A. Postenucleation adjuvant chemotherapy with vincristine, etoposide, and carboplatin for the treatment of high-risk retinoblastoma. Arch. Ophthalmol. 2011, 129, 1422–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, D.H.; Shields, C.L.; Munier, F.L.; Chantada, G.L. Treatment of retinoblastoma in 2015: Agreement and disagreement. JAMA Ophthalmol. 2015, 133, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Fabian, I.D.; Stacey, A.W.; Chowdhury, T.; Duncan, C.; Karaa, E.K.; Scheimberg, I.; Reddy, M.A.; Sagoo, M.S. High-risk histopathology features in primary and secondary enucleated International Intraocular Retinoblastoma Classification Group D eyes. Ophthalmology 2017, 124, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.E.; Shah, S.; Zolfaghari, E.; Jubran, R.; Reid, M.W.; Kim, J.W.; Berry, J.L. An intraocular pressure predictive of high-risk histopathologic features in group E retinoblastoma eyes. Int. Ophthalmol. Clin. 2019, 59, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.W.; Qaddoumi, I.; Billups, C.; Haik, B.G.; Rodriguez-Galindo, C. A clinicopathological correlation of 67 eyes primarily enucleated for advanced intraocular retinoblastoma. Br. J. Ophthalmol. 2011, 95, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Wilson, M.W.; Billups, C.A.; Wu, J.R.; Merchant, T.E.; Brennan, R.C.; Haik, B.G.; Shulkin, B.; Free, T.M.; Given, V.; et al. Pathologic risk-based adjuvant chemotherapy for unilateral retinoblastoma following enucleation. J. Pediatric Hematol. Oncol. 2014, 36, E335–E340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, R.C.; Qaddoumi, I.; Billups, C.A.; Free, T.L.; Haik, B.G.; Rodriguez-Galindo, C.; Wilson, M.W. Comparison of high-risk histopathological features in eyes with primary or secondary enucleation for retinoblastoma. Br. J. Ophthalmol. 2015, 99, 1366–1371. [Google Scholar] [CrossRef]
- Kletke, S.N.; Feng, Z.X.; Hazrati, L.N.; Gallie, B.L.; Soliman, S.E. Clinical predictors at diagnosis of low-risk histopathology in unilateral advanced retinoblastoma. Ophthalmology 2019, 126, 1306–1314. [Google Scholar] [CrossRef]
- Francis, J.H.; Roosipu, N.; Levin, A.M.; Brodie, S.E.; Dunkel, I.J.; Gobin, Y.P.; Abramson, D.H. Current treatment of bilateral retinoblastoma: The impact of intraarterial and intravitreous chemotherapy. Neoplasia 2018, 20, 757–763. [Google Scholar] [CrossRef]
- Abramson, D.H.; Fabius, A.W.; Francis, J.H.; Marr, B.P.; Dunkel, I.J.; Brodie, S.E.; Escuder, A.; Gobin, Y.P. Ophthalmic artery chemosurgery for eyes with advanced retinoblastoma. Ophthalmic Genet. 2017, 38, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.Y.; Dimaras, H.; Massey, C.; Xu, X.L.; Huang, D.S.; Li, B.; Chan, H.S.L.; Gallie, B.L. Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J. Clin. Oncol. 2011, 29, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.E.; Sampor, C.; Solernou, V.; Rossi, J.; Gabri, M.; Eandi-Eberle, S.; de Davila, M.T.G.; Alonso, D.F.; Chantada, G.L. Detection of minimally disseminated disease in the cerebrospinal fluid of children with high-risk retinoblastoma by reverse transcriptase-polymerase chain reaction for GD2 synthase mRNA. Eur. J. Cancer 2013, 49, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.E.; Torbidoni, A.V.; Sampor, C.; Ottaviani, D.; Vazquez, V.; Gabri, M.R.; Garcia de Davila, M.T.; Ramirez-Ortiz, M.A.; Alonso, C.N.; Rossi, J.; et al. Minimal disseminated disease in nonmetastatic retinoblastoma with High-risk pathologic features and association with disease-free survival. JAMA Ophthalmol. 2016, 134, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Hershko, D.D. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008, 112, 1415–1424. [Google Scholar] [CrossRef]
- Brough, R.; Gulati, A.; Haider, S.; Kumar, R.; Campbell, J.; Knudsen, E.; Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Identification of highly penetrant Rb-related synthetic lethal interactions in triple negative breast cancer. Oncogene 2018, 37, 5701–5718. [Google Scholar] [CrossRef] [Green Version]
- Aubry, A.; Yu, T.; Bremner, R. Preclinical studies reveal MLN4924 is a promising new retinoblastoma therapy. Cell Death Discov. 2020, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell. Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef]
- Lough, L.; Sherman, D.; Ni, E.; Young, L.M.; Hao, B.; Cardozo, T. Chemical probes of Skp2-mediated p27 ubiquitylation and degradation. Medchemcomm 2018, 9, 1093–1104. [Google Scholar] [CrossRef]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgard, C.L.; Van Quill, K.R.; O’Brien, J.M. Evaluation of the in vitro and in vivo antitumor activity of histone deacetylase inhibitors for the therapy of retinoblastoma. Clin. Cancer Res. 2008, 14, 3113–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaman-Pillet, N.; Oberson, A.; Schorderet, D.F. BIRO1, a cell-permeable BH3 peptide, promotes mitochondrial fragmentation and death of retinoblastoma cells. Mol. Cancer Res. 2015, 13, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Pasto, G.; Bazan-Peregrino, M.; Olaciregui, N.G.; Restrepo-Perdomo, C.A.; Mato-Berciano, A.; Ottaviani, D.; Weber, K.; Correa, G.; Paco, S.; Vila-Ubach, M.; et al. Therapeutic targeting of the RB1 pathway in retinoblastoma with the oncolytic adenovirus VCN-01. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Berry, J.L.; Xu, L.; Kooi, I.; Murphree, A.L.; Prabakar, R.K.; Reid, M.; Stachelek, K.; Le, B.H.A.; Welter, L.; Reiser, B.J.; et al. Genomic cfDNA analysis of aqueous humor in retinoblastoma predicts eye salvage: The surrogate tumor biopsy for retinoblastoma. Mol. Cancer Res. 2018, 16, 1701–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.L.; Xu, L.; Murphree, A.L.; Krishnan, S.; Stachelek, K.; Zolfaghari, E.; McGovern, K.; Lee, T.C.; Carlsson, A.; Kuhn, P.; et al. Potential of aqueous humor as a surrogate tumor biopsy for retinoblastoma. JAMA Ophthalmol. 2017, 135, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Gerrish, A.; Stone, E.; Clokie, S.; Ainsworth, J.R.; Jenkinson, H.; McCalla, M.; Hitchcott, C.; Colmenero, I.; Allen, S.; Parulekar, M.; et al. Non-invasive diagnosis of retinoblastoma using cell-free DNA from aqueous humour. Br. J. Ophthalmol. 2019, 103, 721–724. [Google Scholar] [CrossRef] [Green Version]
- Kothari, P.; Marass, F.; Yang, J.L.; Stewart, C.M.; Stephens, D.; Patel, J.; Hasan, M.; Jing, X.; Meng, F.; Enriquez, J.; et al. Cell-free DNA profiling in retinoblastoma patients with advanced intraocular disease: An MSKCC experience. Cancer Med. 2020. [Google Scholar] [CrossRef]
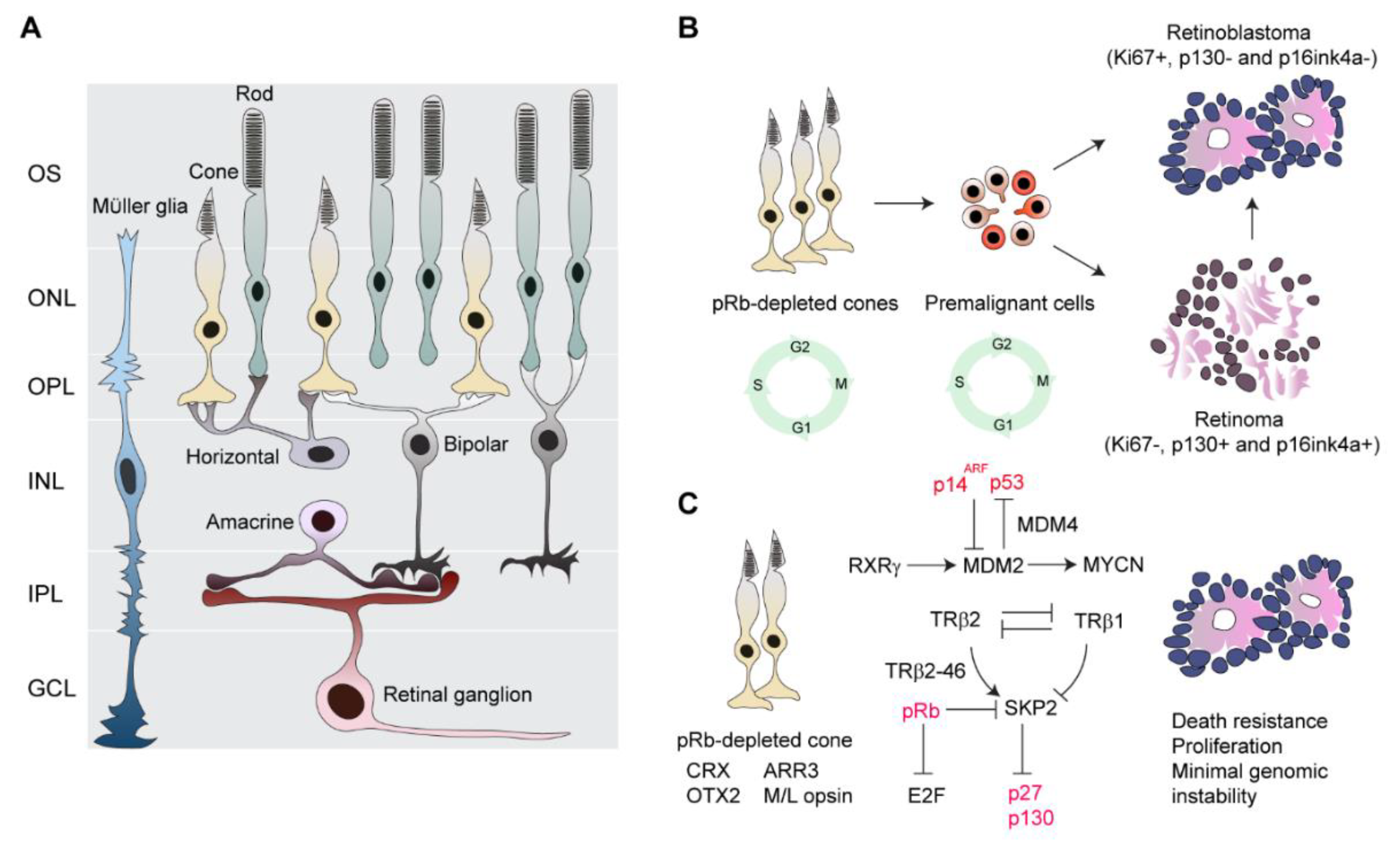

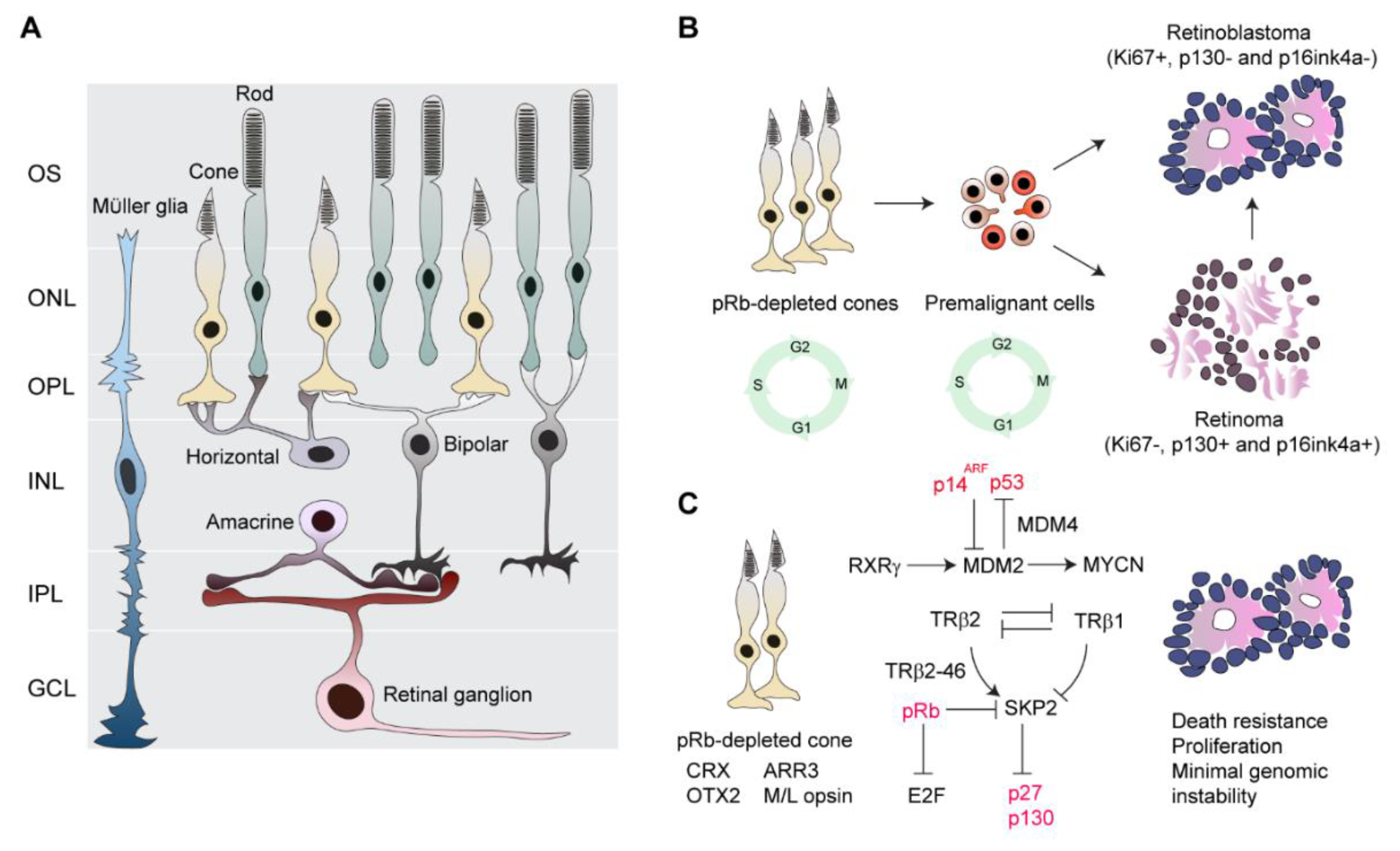{kind=link}
| Reference | 1q Gain | 2p Gain | 6p Gain | 16q Loss | Eye No. | Array |
|---|---|---|---|---|---|---|
| Kooi et al. 2016 [33] | CRB1, NEK7, MIR181 | MYCN | SOX4, DEK | RBL2 | 45 | SNP |
| Mol et al. 2014 [31] | KIF14, MDM4, LRNN2, ZNF281 | MYCN, DDX1 | DEK, E2F3, TNF, KIF13A, TDP2, CAP2, NUP153, SOX4, ID4 | CDH11, CDH13, RBL2, MBTPS1, ZCCHC14, ZDHHC7 | 21 | SNP |
| Sampieri et al. 2009 [4] | MCL1, SHC1, MUC1 | MYCN, DDX1 | IRF4, DEK, PIM1, E2F3, CCND3 | CYLD, RBL2 | 18 | CGH |
| Zielinski et al. 2005 [32] | SHC1, MDM4, GAC1 | MYCN | TNF-alpha, HLA gene cluster | RBL2 | 17 | CGH |
| Chen et al. 2001 [27] | LRNN2, REN, GAC1 | MYCN | E2F3, ID4 | CALB1, CBFB, CDH1, CDH11, CDH13, CDH15, CDH16, CDH3, CDH5, CDH8, E2F4, MAF, ZFHX3 | 50 | CGH |
| Herzog et al. 2001 [28] | MYCN | RBL2 | 26 | CGH |
| Clinicopathological Characteristics | Genomic Instability | Photoreceptor Gene Signature | |
|---|---|---|---|
| High | Low | ||
| Laterality [33,45] | Unilateral | Bilateral | Unilateral |
| Tumor grade * [33,44,45] | Less differentiation | Differentiation | Less differentiation |
| Age at diagnosis/enucleation [33,45,51,52] | Older | Younger | Older |
| High-risk features (optic nerve/choroid invasion) [33,44,45] | Not associated | Not associated | Not associated |
| Tumor volume [33,45] | Not associated | Smaller | Larger |
| Tumor location [45,53] | N/a | Central retina | Peripheral retina/entire retina |
| Eye size [53] | N/a | Smaller | Larger |
| Number of lesions [33,53] | Not associated | 1–5 lesions | >5 lesions |
| Chemotherapy sensitivity [45] | N/a | Less sensitivity | More sensitivity |
| Genotype of the Genetically Engineered Mouse Model | Characteristics | Study Objective |
|---|---|---|
| Rb1/p107 DKO [50,88,90] | 68% of mice with RB develop by 280 days and 22% of mice carry bilateral RB | Additional genes required in cooperation with pRb loss for tumorigenesis and early RB |
| Rb1/p130 DKO [50,85,88] | 85% of mice with RB develop by 128 days and 28% of mice carry bilateral RB | Early and advanced RB |
| Rb1/p107/p130 TKO [50] | 100% of mice with RB develop by 80 days and 83% mice of carry bilateral RB | Very aggressive RB |
| Rb1/p107/p53 TKO [50,90] | 98–100% of mice with RB develop by 100 days and 65% of mice carry bilateral RB | Advanced and aggressive RB |
| Rb1/p107 DKO/MDMX Tg [50] | 90% of mice with RB and 63% of mice carry bilateral RB | Advanced and aggressive RB |
| Rb1/p107/Pten TKO [93] | 100% of mice with bilateral RB develop by 30 days | Tumor progression related to the PI3K/AKT pathway |
| Rb1 KO/MYCN [83] | 100% of mice with RB develop by 54 days | Oncogenic effects of MYCN on RB |
| Histopathological Features at Primary Enucleation (% Enucleated Eyes with Risk Features) | Total Eye No. | Correlated Clinical Features (% Patients with Described Features Exhibiting Clinicopathological Correlations) |
|---|---|---|
| LR (89.5%) * [149] | 38 | Macular spare (26%) Optic nerve visibility (42%) <1 quadrant of retinal detachment (22%) (studied in group D eyes) |
| HR (47%) [145] | 96 | Raised intraocular pressure (>34 mmHg) in group E eyes (100%) |
| HR (12.5%) [144] | 40 | No vitreous seeds in group D eyes (20%) |
| HR (36%) [115] | 403 | Group E eyes (39%) Delayed time to treatment >6 months (63%) Secondary glaucoma (64%) |
| HR (23%) [114] | 519 | Group D (17%) and E (24%) eyes |
| HR (64%) [52] | 76 | Iris neovascularization (63%) Raised intraocular pressure (>21 mmHg) (63%) ** Shallow anterior chamber (26%) (studied in group E eyes) |
| HR (25%) [146] | 67 | Group E eyes (50%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewkhaw, R.; Rojanaporn, D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers 2020, 12, 2304. https://doi.org/10.3390/cancers12082304
Kaewkhaw R, Rojanaporn D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers. 2020; 12(8):2304. https://doi.org/10.3390/cancers12082304
Chicago/Turabian StyleKaewkhaw, Rossukon, and Duangnate Rojanaporn. 2020. "Retinoblastoma: Etiology, Modeling, and Treatment" Cancers 12, no. 8: 2304. https://doi.org/10.3390/cancers12082304
APA StyleKaewkhaw, R., & Rojanaporn, D. (2020). Retinoblastoma: Etiology, Modeling, and Treatment. Cancers, 12(8), 2304. https://doi.org/10.3390/cancers12082304