Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
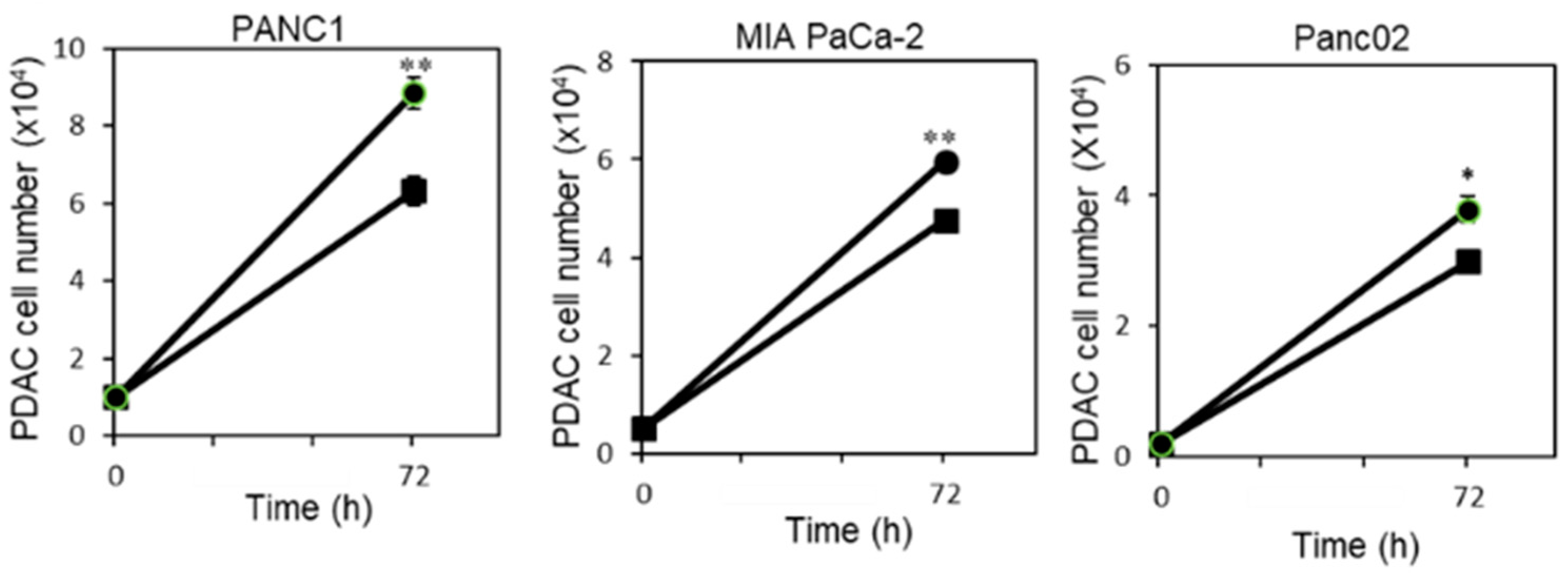
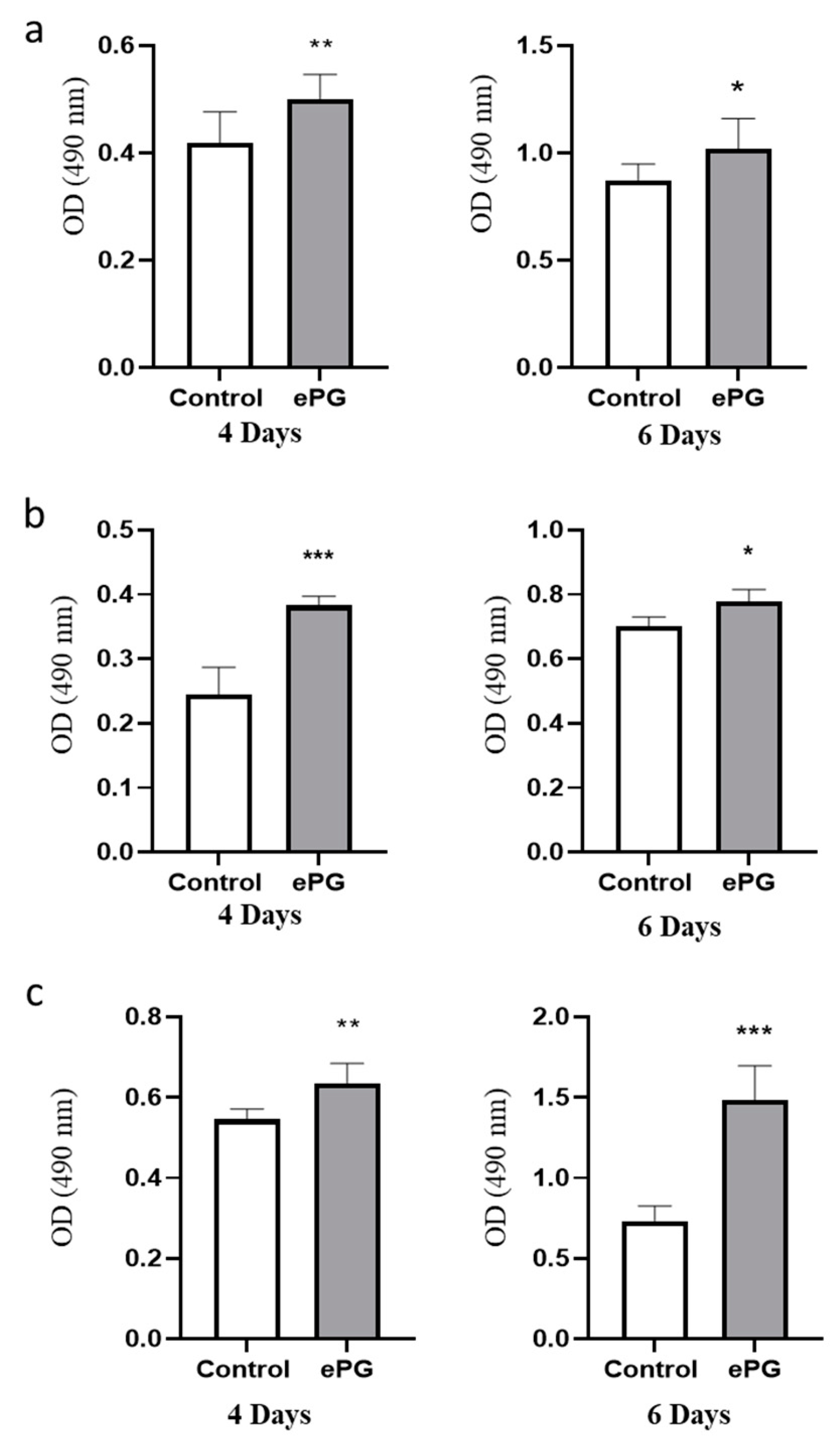
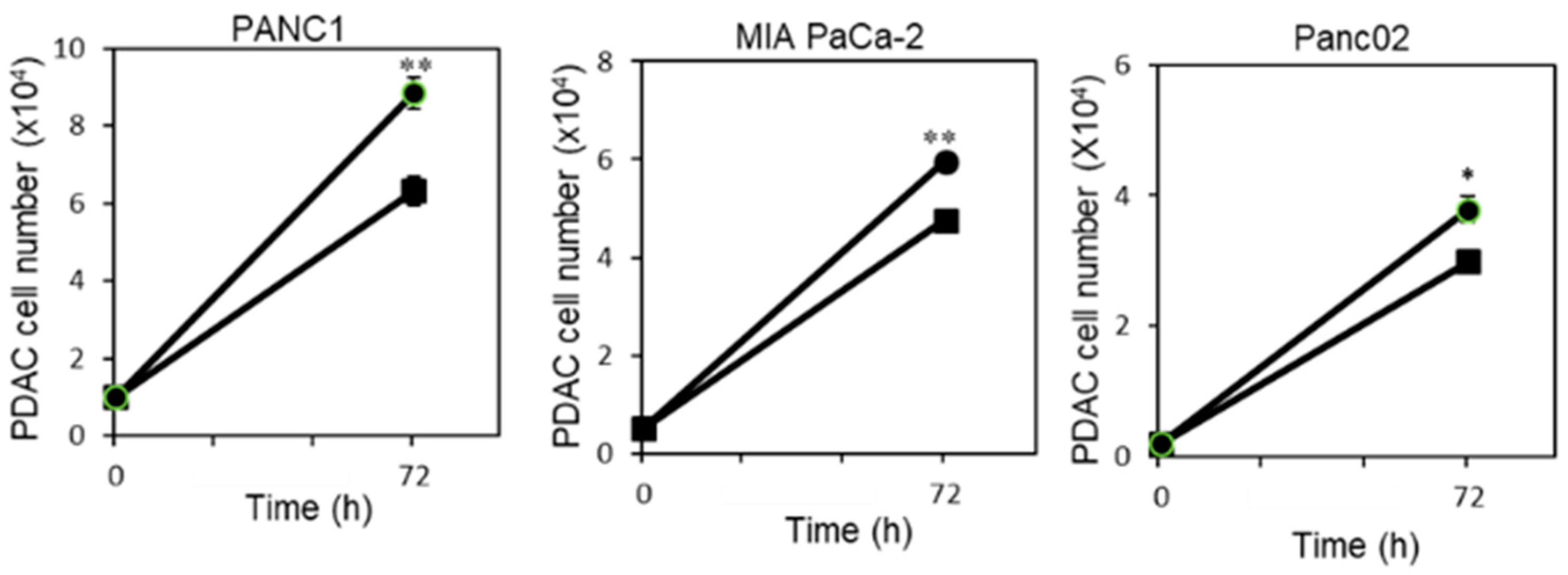
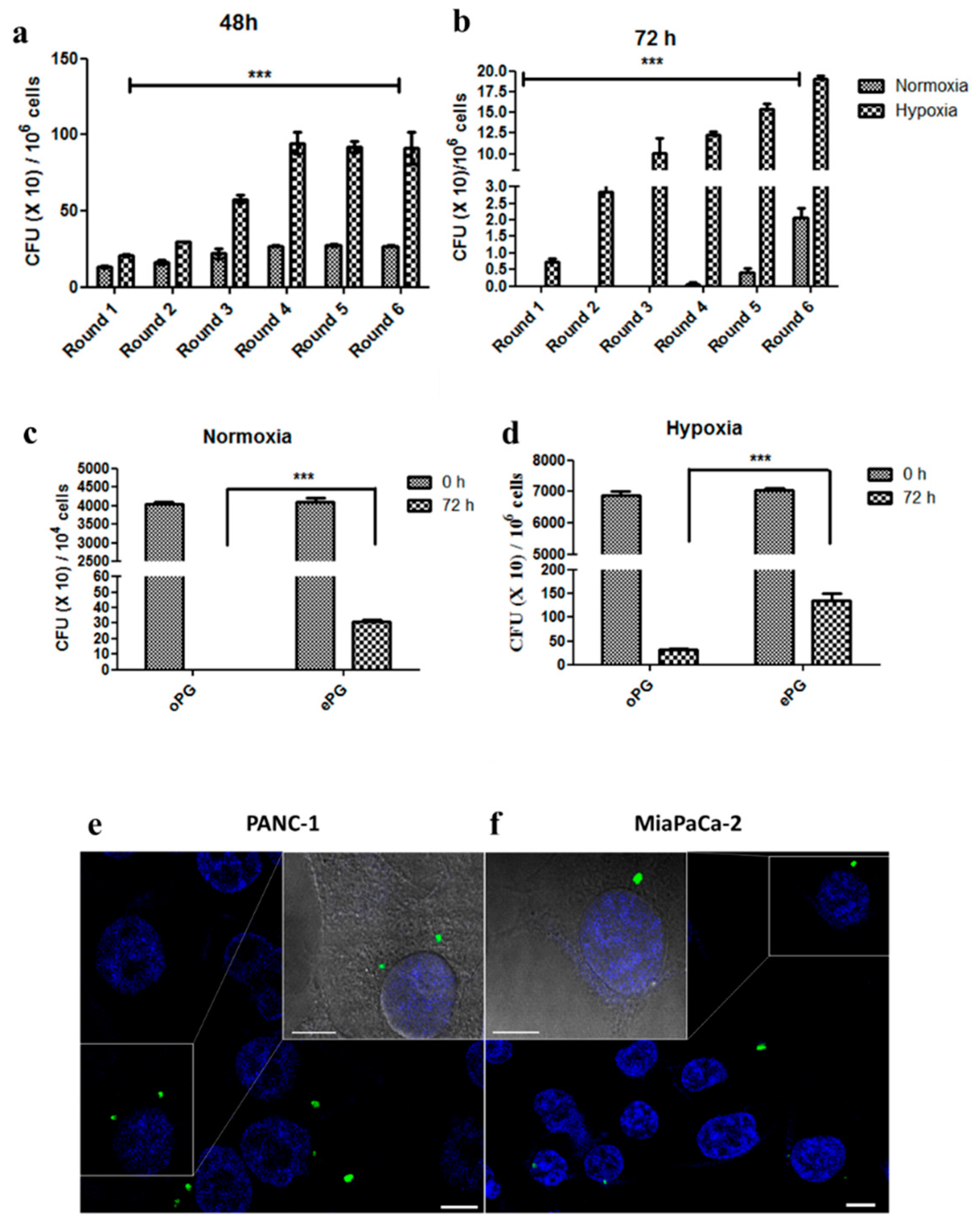
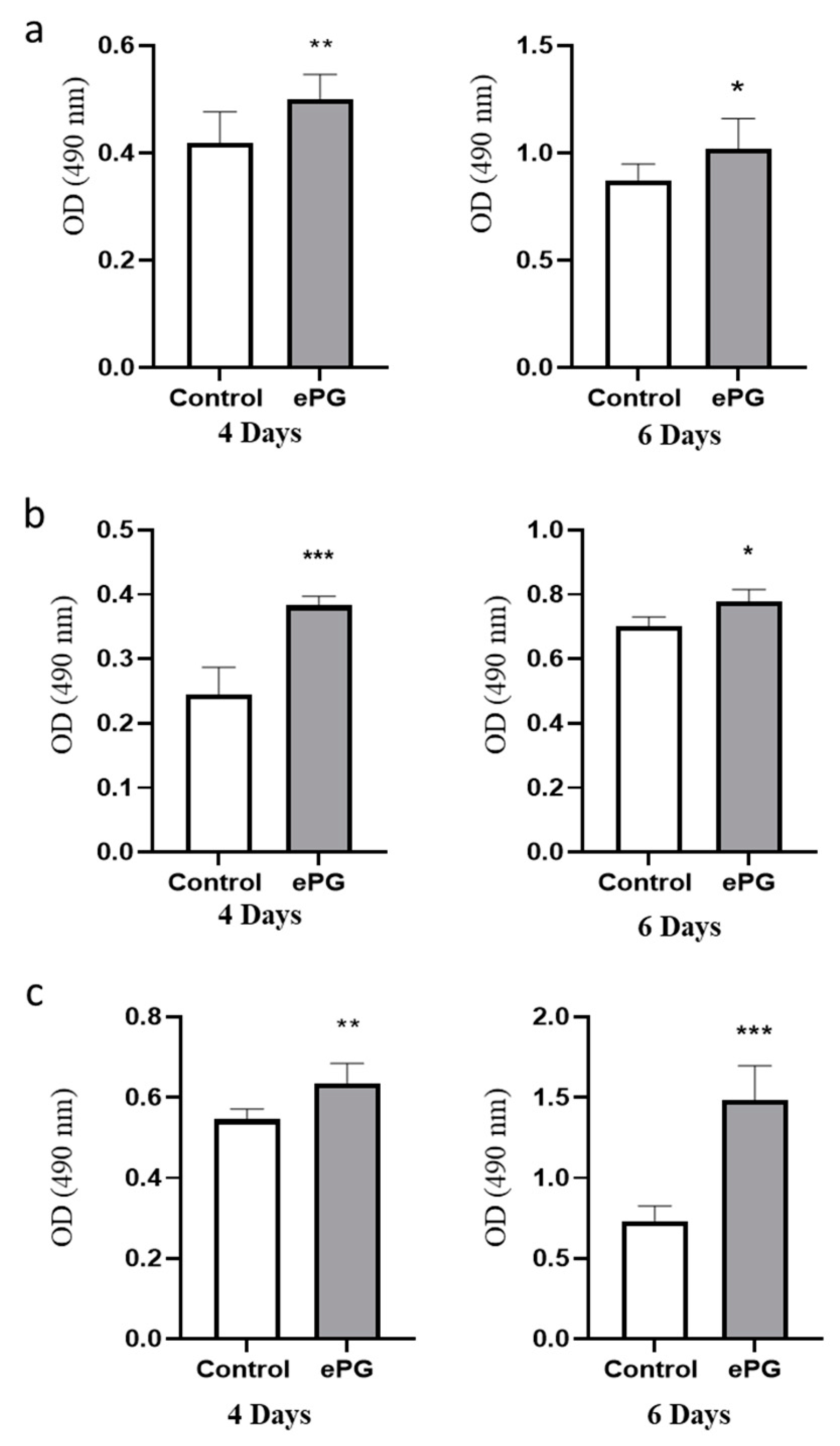
2.1. P. gingivalis Infection Enhances Pancreatic Tumor Cell Proliferation
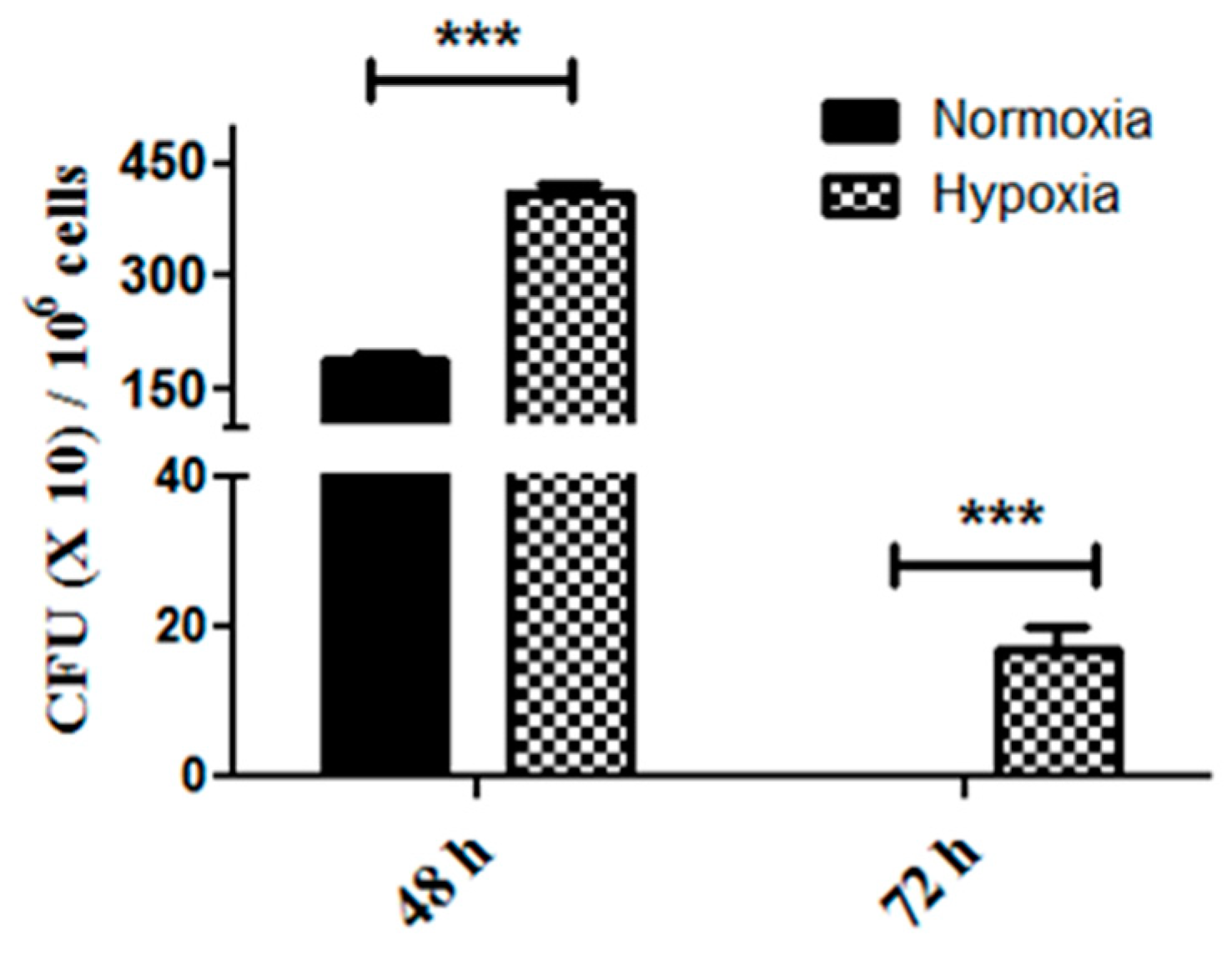
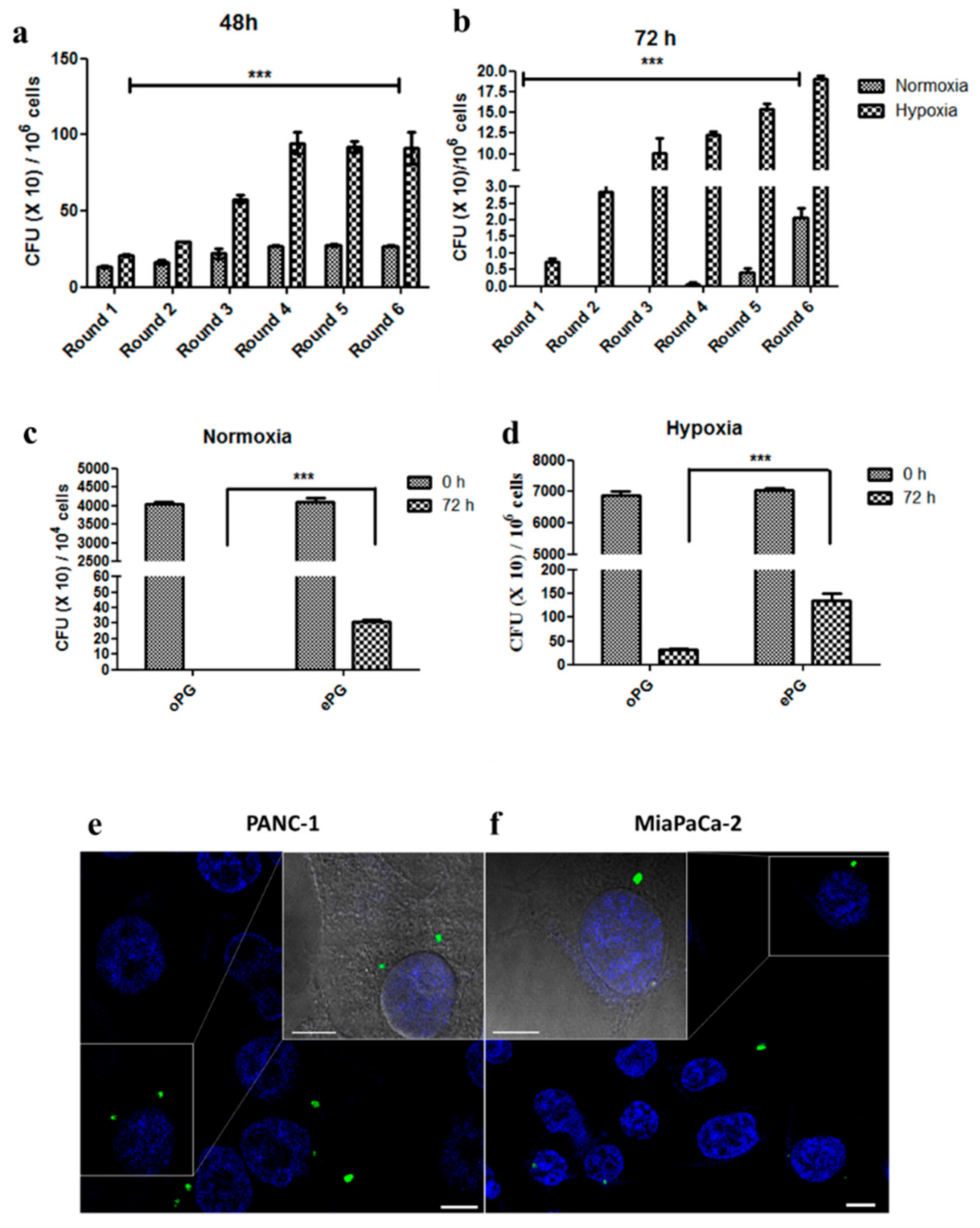
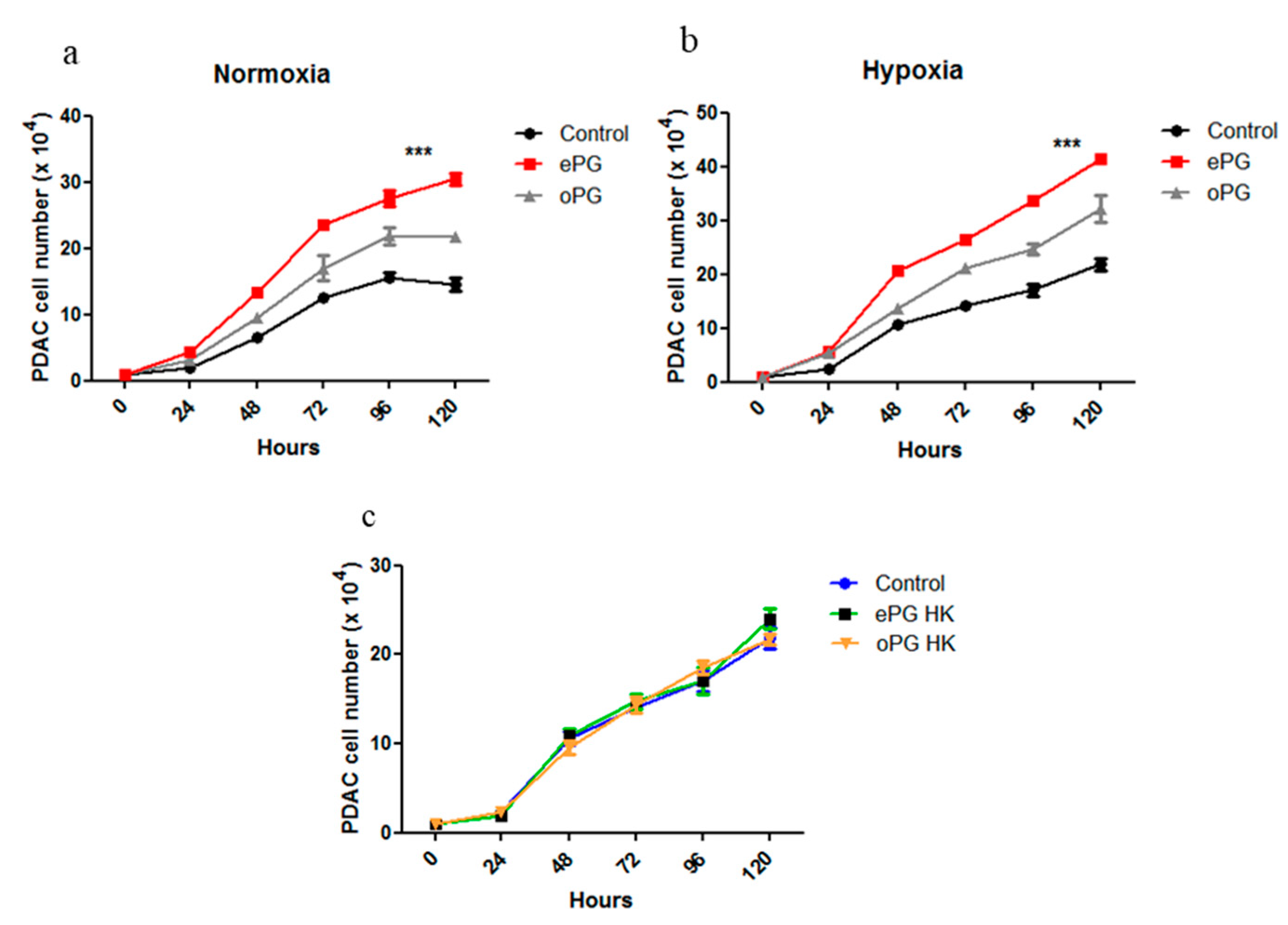
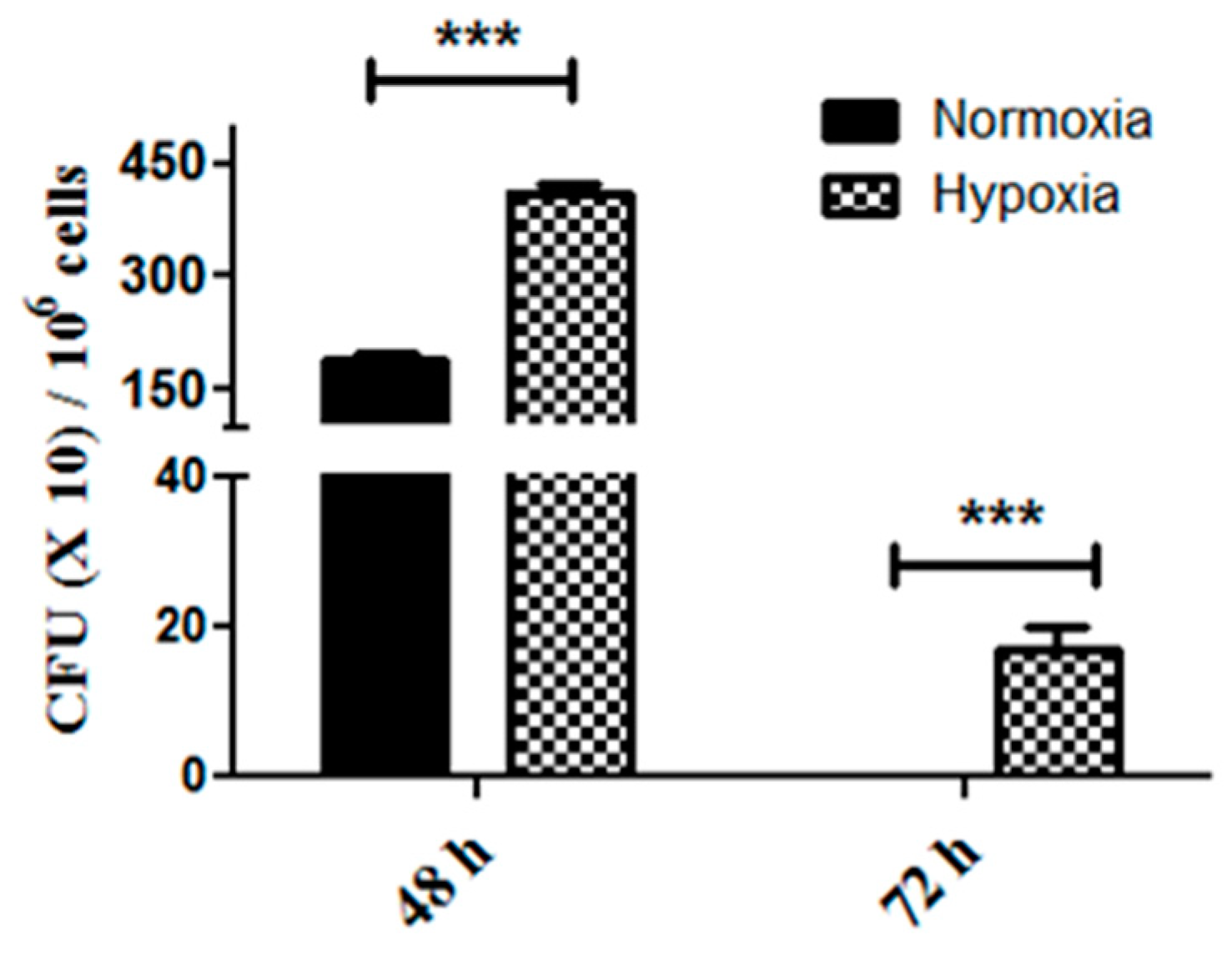
2.2. Enhanced Tumor Cell Proliferation Correlates with P. gingivalis Intracellular Survival
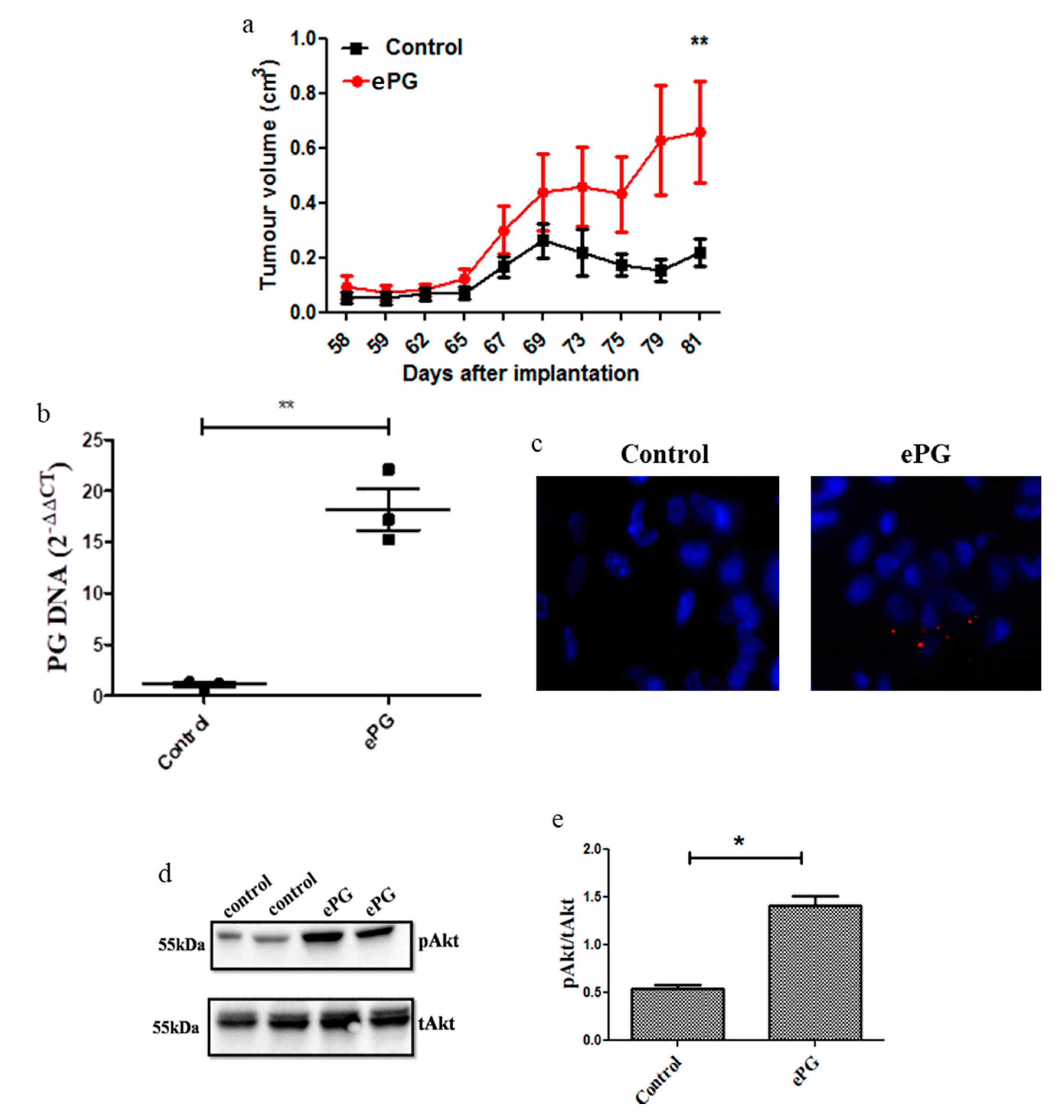
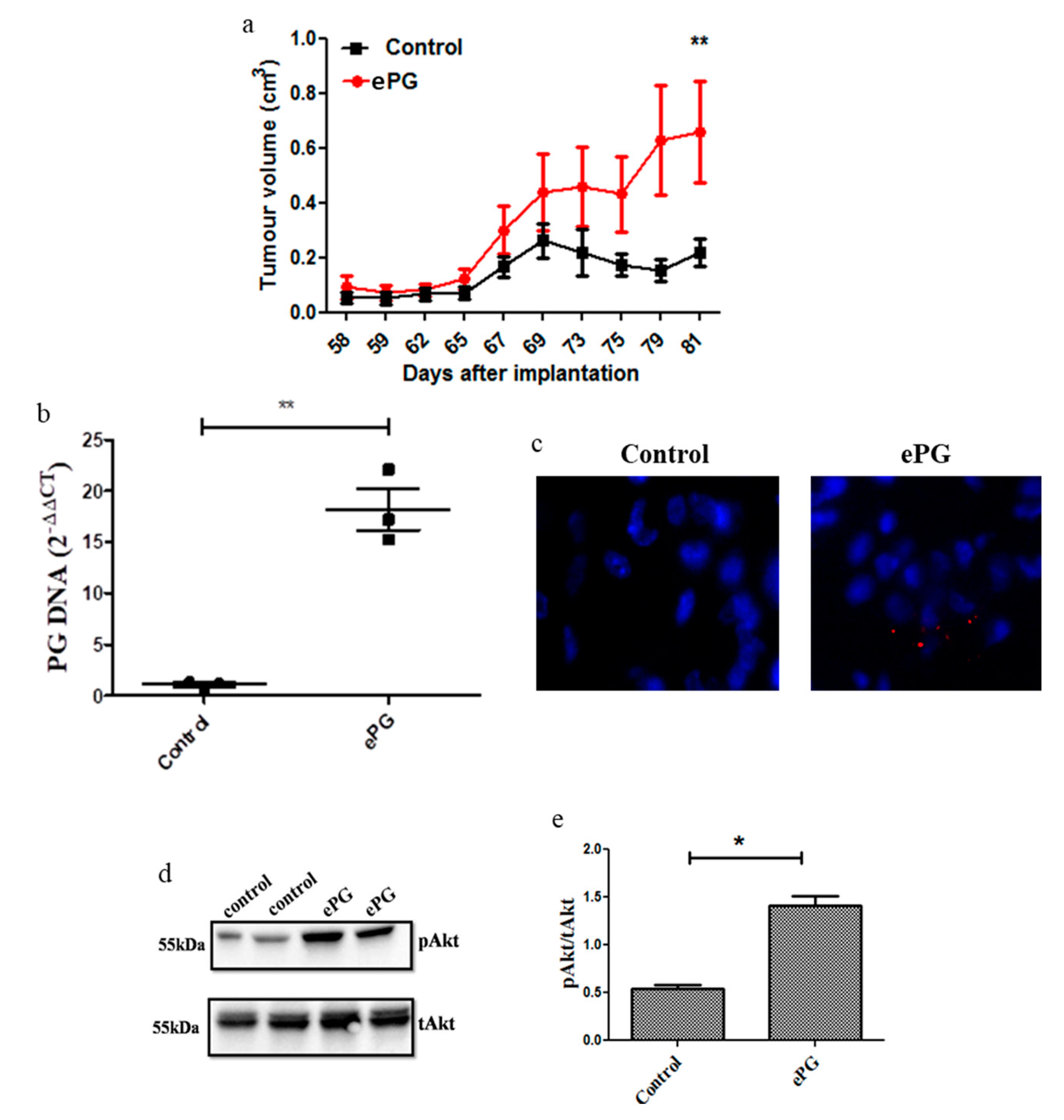
2.3. P. gingivalis Enhances Tumor Growth and Akt Activation
3. Discussion
4. Materials and Methods
4.1. Bacteria and Growth Conditions
4.2. Cell Lines and Culture Conditions
4.3. Cell Proliferation Assay
4.4. P. gingivalis Intracellular Survival Assay (ICS)
4.5. Detection of Intracellular P. gingivalis
4.6. Analysis of Gene Expression by qRT-PCR
4.7. Tumor Growth In Vivo
4.8. Detection of P. gingivalis in Tumor Tissue
4.9. Fluorescence In Situ Hybridization (FISH)
4.10. Western Blot
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Regel, I.; Mayerle, J.; Mahajan, U.M. Current strategies and future perspectives for precision medicine in pancreatic cancer. Cancers 2020, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Segers, S.; Hayes, R.B. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis 2012, 33, 1055–1058. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, D.S.; Izard, J.; Wilhelm-Benartzi, C.S.; You, D.H.; Grote, V.A.; Tjonneland, A.; Dahm, C.C.; Overvad, K.; Jenab, M.; Fedirko, V.; et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013, 62, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Makkawi, H.; Hoch, S.; Burns, E.; Hosur, K.; Hajishengallis, G.; Kirschning, C.J.; Nussbaum, G. Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front. Cell. Infect. Microbiol. 2017, 7, 359. [Google Scholar] [CrossRef]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Olsen, I.; Progulske-Fox, A. Invasion of Porphyromonas gingivalis strains into vascular cells and tissue. J. Oral Microbiol. 2015, 7, 28788. [Google Scholar] [CrossRef] [Green Version]
- Olsen, I.; Yilmaz, O. Possible role of Porphyromonas gingivalis in orodigestive cancers. J. Oral Microbiol. 2019, 11, 1563410. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Yoshida, K.; Okamura, H.; Ochiai, K.; Takamura, H.; Fujiwara, N.; Ozaki, K. Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the Akt/GSK-3beta signaling pathway. Biochim. Biophys. Acta 2013, 1832, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.; Chegini, N.; Shiverick, K.T.; Lamont, R.J. Localization of P. gingivalis in preterm delivery placenta. J. Dent. Res. 2009, 88, 575–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilievski, V.; Toth, P.T.; Valyi-Nagy, K.; Valyi-Nagy, T.; Green, S.J.; Marattil, R.S.; Aljewari, H.W.; Wicksteed, B.; O’Brien-Simpson, N.M.; Reynolds, E.C.; et al. Identification of a periodontal pathogen and bihormonal cells in pancreatic islets of humans and a mouse model of periodontitis. Sci. Rep. 2020, 10, 9976. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Baque, V.; Garidou, L.; Pomie, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, N.; Marin, M.J.; Laguna, E.; Herrera, D.; Sanz, M.; Figuero, E. Detection and quantification of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in bacteremia induced by interdental brushing in periodontally healthy and periodontitis patients. Arch. Oral Biol. 2019, 98, 213–219. [Google Scholar] [CrossRef]
- Liu, X.B.; Gao, Z.Y.; Sun, C.T.; Wen, H.; Gao, B.; Li, S.B.; Tong, Q. The potential role of P.gingivalis in gastrointestinal cancer: A mini review. Infect. Agents Cancer 2019, 14, 23. [Google Scholar] [CrossRef]
- Vaz, J.; Andersson, R. Intervention on toll-like receptors in pancreatic cancer. World J. Gastroenterol. 2014, 20, 5808–5817. [Google Scholar] [CrossRef]
- Gallimidi, A.B.; Fischman, S.; Revach, B.; Bulvik, R.; Maliutina, A.; Rubinstein, A.M.; Nussbaum, G.; Elkin, M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 2015, 6, 22613–22623. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, S.E.; Lamont, R.J. Oral bacteria and cancer. PLoS Pathog. 2014, 10, e1003933. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Roberts, J.S.; Choi, C.H.; Atanasova, K.R.; Yilmaz, O. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 2018, 9, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Gohar, A.; Zarei, M.; Brody, J.R.; Winter, J.M. Metabolic dependencies in pancreatic cancer. Front. Oncol. 2018, 8, 617. [Google Scholar] [CrossRef]
- Goto, Y.; Arigami, T.; Kitago, M.; Nguyen, S.L.; Narita, N.; Ferrone, S.; Morton, D.L.; Irie, R.F.; Hoon, D.S. Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol. Cancer Ther. 2008, 7, 3642–3653. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, O.; Verbeke, P.; Lamont, R.J.; Ojcius, D.M. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect. Immun. 2006, 74, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Kornmann, M.; Ishiwata, T.; Itakura, J.; Tangvoranuntakul, P.; Beger, H.G.; Korc, M. Increased cyclin D1 in human pancreatic cancer is associated with decreased postoperative survival. Oncology 1998, 55, 363–369. [Google Scholar] [CrossRef]
- Hermano, E.; Meirovitz, A.; Meir, K.; Nussbaum, G.; Appelbaum, L.; Peretz, T.; Elkin, M. Macrophage polarization in pancreatic carcinoma: Role of heparanase enzyme. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef]
- Sunde, P.T.; Olsen, I.; Gobel, U.B.; Theegarten, D.; Winter, S.; Debelian, G.J.; Tronstad, L.; Moter, A. Fluorescence in situ hybridization (FISH) for direct visualization of bacteria in periapical lesions of asymptomatic root-filled teeth. Microbiology 2003, 149, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, O.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 2004, 72, 3743–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuboniwa, M.; Hasegawa, Y.; Mao, S.; Shizukuishi, S.; Amano, A.; Lamont, R.J.; Yilmaz, O.P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 2008, 10, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [Green Version]
- Tribble, G.D.; Lamont, R.J. Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontol. 2000 2010, 52, 68–83. [Google Scholar] [CrossRef]
- Madianos, P.N.; Papapanou, P.N.; Nannmark, U.; Dahlen, G.; Sandros, J. Porphyromonas gingivalis FDC381 multiplies and persists within human oral epithelial cells in vitro. Infect. Immun. 1996, 64, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Boutin, A.T.; Zinkernagel, A.S.; Datta, V.; Nizet, V.; Johnson, R.S. Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J. Investig. Dermatol. 2008, 128, 1964–1968. [Google Scholar] [CrossRef] [Green Version]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 2001, 200, 145–149. [Google Scholar] [CrossRef]
- Soto, C.; Bugueno, I.; Hoare, A.; Gonzalez, S.; Venegas, D.; Salinas, D.; Melgar-Rodriguez, S.; Vernal, R.; Gamonal, J.; Quest, A.F.; et al. The Porphyromonas gingivalis O antigen is required for inhibition of apoptosis in gingival epithelial cells following bacterial infection. J. Periodontal. Res. 2016, 51, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, J.; Lin, L.; Zhao, H.; Miao, L.; Pan, Y. Porphyromonas gingivalis degrades integrin beta1 and induces AIF-mediated apoptosis of epithelial cells. Infect. Dis. (Lond.) 2019, 51, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, R.; Yi, Z.; Li, Y.; Fu, Y.; Zhang, Y.; Li, P.; Li, X.; Pan, Y. Porphyromonas gingivalis induced inflammatory responses and promoted apoptosis in lung epithelial cells infected with H1N1 via the Bcl2/Bax/Caspase3 signaling pathway. Mol. Med. Rep. 2018, 18, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Lu, S.; Wei, M.; Cai, X.; Wang, G. Identification of novel genes involved in gingival epithelial cells responding to Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis infections. Arch. Oral Biol. 2018, 96, 113–121. [Google Scholar] [CrossRef]
- Bold, R.J.; Virudachalam, S.; McConkey, D.J. BCL2 expression correlates with metastatic potential in pancreatic cancer cell lines. Cancer 2001, 92, 1122–1129. [Google Scholar] [CrossRef]
- Murthy, D.; Attri, K.S.; Singh, P.K. Phosphoinositide 3-Kinase signaling pathway in pancreatic ductal adenocarcinoma progression, pathogenesis, and therapeutics. Front. Physiol. 2018, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, P.H.; Reyes, L.; Chadda, A.S.; Belanger, M.; Wallet, S.M.; Akin, D.; Dunn, W., Jr.; Progulske-Fox, A. Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: A comparative study. PLoS ONE 2012, 7, e52606. [Google Scholar] [CrossRef]
- Tanner, A.C.; Haffer, C.; Bratthall, G.T.; Visconti, R.A.; Socransky, S.S. A study of the bacteria associated with advancing periodontitis in man. J. Clin. Periodontol. 1979, 6, 278–307. [Google Scholar] [CrossRef]
- Ilievski, V.; Bhat, U.G.; Suleiman-Ata, S.; Bauer, B.A.; Toth, P.T.; Olson, S.T.; Unterman, T.G.; Watanabe, K. Oral application of a periodontal pathogen impacts SerpinE1 expression and pancreatic islet architecture in prediabetes. J. Periodontal. Res. 2017, 52, 1032–1041. [Google Scholar] [CrossRef]
- Kiss, B.; Miko, E.; Sebo, E.; Toth, J.; Ujlaki, G.; Szabo, J.; Uray, K.; Bai, P.; Arkosy, P. Oncobiosis and microbial metabolite signaling in pancreatic adenocarcinoma. Cancers 2020, 12, 1068. [Google Scholar] [CrossRef]
- Bauer, C.; Bauernfeind, F.; Sterzik, A.; Orban, M.; Schnurr, M.; Lehr, H.A.; Endres, S.; Eigler, A.; Dauer, M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut 2007, 56, 1275–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnanasekaran, J.; Binder Gallimidi, A.; Saba, E.; Pandi, K.; Eli Berchoer, L.; Hermano, E.; Angabo, S.; Makkawi, H.; Khashan, A.; Daoud, A.; et al. Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers 2020, 12, 2331. https://doi.org/10.3390/cancers12082331
Gnanasekaran J, Binder Gallimidi A, Saba E, Pandi K, Eli Berchoer L, Hermano E, Angabo S, Makkawi H, Khashan A, Daoud A, et al. Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers. 2020; 12(8):2331. https://doi.org/10.3390/cancers12082331
Chicago/Turabian StyleGnanasekaran, JebaMercy, Adi Binder Gallimidi, Elias Saba, Karthikeyan Pandi, Luba Eli Berchoer, Esther Hermano, Sarah Angabo, Hasna′a Makkawi, Arin Khashan, Alaa Daoud, and et al. 2020. "Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells" Cancers 12, no. 8: 2331. https://doi.org/10.3390/cancers12082331
APA StyleGnanasekaran, J., Binder Gallimidi, A., Saba, E., Pandi, K., Eli Berchoer, L., Hermano, E., Angabo, S., Makkawi, H., Khashan, A., Daoud, A., Elkin, M., & Nussbaum, G. (2020). Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers, 12(8), 2331. https://doi.org/10.3390/cancers12082331