1. Introduction
Neuroblastoma (NB) is a common extracranial solid tumor of the developing sympathetic nervous system, which accounts for roughly 5% of all diagnosed pediatric cancers [
1]. Patients with localized tumors, defined as low or intermediate risk, do not require intensive therapeutic treatment, as, in most cases, the tumor regresses spontaneously. For such patients, surgery is performed when possible and chemotherapy is only considered for symptomatic tumors or for tumor masses growing after surgery [
1,
2]. On the contrary, patients with disseminated tumors, defined as high-risk, undergo intensive treatment that includes different phases: induction based on chemotherapy at maximally tolerated doses, local treatment with surgery and radiotherapy, consolidation with high dose chemotherapy and peripheral blood stem cells rescue, maintenance based on a differentiating agent (cis-retinoic acid) and, more recently, on immunotherapy targeting the expression of disialoganglioside (GD2) on NB cells. Despite this aggressive treatment, almost 50% of high-risk NB patients are refractory to therapy, relapse, and die [
1].
Efforts to identify prognostic biomarkers from the genomic interrogation of NB tumors have been made with the aim of improving patient stratification and providing novel therapeutic targets [
1,
2]. Molecular signatures are becoming increasingly important tools for assisting clinicians in prognosis assessment and therapeutic decisions, because they can be used for accurately predicting patient outcome, relapse, or response to therapy, and also be instrumental for refining patient risk stratification, optimizing treatment, and reducing unnecessary therapy related toxicity [
3,
4,
5,
6]. For these purposes, the gene expression profiles of a large number of primary tumor specimens of NB patients have been published in distinct data sets becoming available to the scientific community [
6,
7,
8,
9,
10,
11,
12,
13]. However, a large-scale expression study of NB tumors has not been previously carried out because different technologies use different proprietary annotations to identify transcripts. The integration of the distinct data sets into one merged data set would enable an unbiased and robust prediction analysis because data set may be split into two large training and test sets. As a consequence, the availability of a large number of gene expression profiles coupled with patient characteristics may enable the stratification of subgroups of patients that are notoriously difficult to analyze with a low number of cases.
The importance of hypoxia in conditioning the aggressiveness of tumors, including NB, is documented by an extensive literature [
14,
15,
16,
17]. A variety of techniques have been described to measure intratumor hypoxia including polarographic electrodes, fiber-optic probes, and positron emission tomography, but there is no consensus on the most appropriate approach to use [
18]. The identification of an accurate hypoxia predictor may be instrumental for discriminating diagnosis patients who will potentially benefit of an anti-hypoxia therapy, thus preventing treatment-associated damage elicited by unnecessary therapies. We have previously used a biology-driven approach to assess the hypoxic status of NB tumors, consisting in the analysis of the gene expression profile of NB cell lines cultured under hypoxic and normoxic conditions, and identified an 11-probe set hypoxia signature that was able to accurately predict NB patient outcome [
19]. However, application of this signature in a large multiplatform study in NB has never been evaluated.
The tumor microenvironment (TME) is a heterogeneous milieu that is composed by neoplastic, stromal, endothelial, and infiltrating immune cells [
20]. The functional interaction among different TME components is critical to determine the development and progression of several types of cancers, including NB [
21,
22]. Unmasking the altered molecular mechanisms in a hypoxic NB TME may be instrumental to identify novel therapeutic targets and pathways that are involved in NB tumor progression and to design novel personalized therapies for NB patients who have low probability to survive with actual treatment strategies. Hypoxia was reported to strongly affect the TME by altering important biological processes, including tumor cell differentiation, survival, migration, and resistance to therapy, and influencing the nature and function of the immune cell infiltrate [
14,
23]. However, the molecular mechanisms and biological effectors that are involved in NB hypoxic TME have been only partially elucidated.
Telomere maintenance mechanisms (TMM) are adopted by tumor cells to prevent telomere shortening and acquire immortality and they represent a malignant hallmark of several cancer cells [
24]. Telomerase is a complex ribonucleic reverse transcriptase that is responsible for telomere maintenance by synthesizing telomeric DNA repeats at the 3′ ends of linear chromosomes [
24]. The catalytic subunit of the human telomerase reverse transcriptase (TERT) is a key component of the telomerase complex and it is detectable in over 90% of human cancers [
25]. Alternative lengthening of telomeres (ALT) is an intra-telomeric recombination mechanism that may be employed by tumor cells to maintain telomere lengthening independently by telomerase activation [
24]. Alterations found to be responsible for TMM in tumor cells include TERT rearrangement, somatic mutations of the TERT promoter, ALT, epigenetic changes, and amplification of TERT gene [
24]. Despite the large number of publications reporting the critical role of TMM in different diseases, the mechanisms of telomerase regulation remain mostly unknown [
26,
27]. Ackermann and coworkers have recently shown the unfavorable prognostic impact of TMM in combination with RAS and/or p53 pathway mutations in NB and the correlation of high expression levels of the TERT gene with TMM in a large set of NB specimens [
13].
In this study, we aimed at assessing the role of hypoxia in the pathogenesis of NB by analyzing the molecular mechanisms and biological effectors that are involved in NB hypoxic TME and at dissecting the prognostic value of a new hypoxia-based predictor in a large multicenter and multiplatform study. Our results show the unfavorable prognostic value of hypoxia in a large number of patients and the ability of hypoxia to additionally stratify clinically relevant groups of NB patients. Furthermore, our results reveal the deregulation of specific biological processes and pathways affecting NB TME.
2. Results
2.1. Collection of the Gene Expression Profile of NB Primary Tumors and Patient Characteristics
Analyses were carried out using the 11-probe set signature to assess the role of hypoxia in the pathogenesis of NB and to dissect the prognostic value of a new hypoxia-based predictor in a large multicenter and multiplatform study [
19]. To these aims, we collected the gene expression profile of 1882 NB tumor specimens covering the entire spectrum of the disease included into four publicly available data sets (RNA-seq498, Affymetrix413, Agilent709, and Agilent262) [
6,
7,
8,
9,
10,
11,
12,
13].
Figure 1 shows the schematic representation of the analyses carried out in the present study.
The relative percentage of patient outcome was comparable among the four data sets.
Table 1 summarizes platform information, clinical, and molecular characteristics of patients.
Age at diagnosis, international NB staging system (INSS) stage, MYCN status, event overall, and event-free data were available for all data sets. Patient follow-up was available for RNA-seq498, Affymetrix413, and Agilent709 data sets, but not for Agilent262, whereas telomere maintenance, ALT, documented regression, TERT rearrangement, ATRX mutation, RAS, and p53 mutation data were only available for the Agilent262 data set.
2.2. Integration of Gene Expression Profiles of NB Primary Tumors Using the COMBAT Batch-Effect Removal Method
The gene expression profiles of RNA-seq498, Agilent709, and Affymetrix413 data sets were integrated into a single data set to achieve a large-scale genomic data analysis. 577 patients were filtered out from the analysis either because of missing information about outcome or of follow-up shorter than five years (
Figure 1). Furthermore, 257 patients that were profiled with both Illumina and Agilent technologies were removed from the Agilent709 data set to have independent data sets. Proprietary identifiers of each platform were mapped into gene symbols for comparability. The new merged and filtered data set comprised 786 patients that included 288 patients from Agilent709, 367 from RNA-seq498 and 131 from Affymetrix413.
It is known that the batch effect may be introduced when data sets from different gene expression platforms are integrated [
28]. Batch effect can be estimated using principal variance component analysis (PVCA). PVCA uses the weighted average proportion variance (WAPV) to estimate the magnitude of any source of variability using biological, clinical, and batch variables [
28]. Thus, the presence of a potential batch effect in the merged data set was assessed by PVCA. The PVCA results showed a WAPV of platform of 0.79, indicating that data integration introduced a measurable batch effect (
Figure S1). Several computational methods have been proposed to remove batch effect from the data [
28]. COMBAT is a well-known method to remove batch effect in the data applying an empirical bayes approach [
28]. The application of the COMBAT technique to the gene expression profiles of RNA-seq498, Agilent709 and Affymetrix413 data sets removed the batch effect introduced by integrating the data from three different platforms (WAPV of Platform = 0.0;
Figure S2). Furthermore, analysis evidenced the variance explained by the biological and clinical variables (WAPV > 0,
Figure S2). The 786 gene expression profiles of the batch-adjusted data set are available in
Table S1.
The expression profiles of 236 randomly selected tumors out of 786 (30%) served to build a classifier and the profiles of the remaining 550 tumors (70%) were used to test its prognostic value in a validation data set. The clinical and molecular characteristics of patients in the training and test sets are summarized in
Table S2 and listed in
Table S3.
2.3. Identification of a New Multiplatform Hypoxia Biomarker
We have previously used a biology-driven approach in order to assess the hypoxic status of NB cells identifying a 11 probe set signature that was able to accurately predict NB patient outcome [
19]. This signature could not be used in the present study because the probe identifiers used were specific of the Affymetrix U133 plus 2.0 gene expression platform. Hence, we refined the signature by mapping the probe sets into gene symbols to obtain a multi-platform biomarker. 9 out of the 11 probe sets were annotated with a gene symbol, whereas two were not associated with a gene symbol and were excluded from subsequent analysis. Seven out of nine gene symbols were unique. Therefore, a new seven-gene biomarker named NB-hop (NB-hypoxia outcome prediction) was defined.
Table 2 lists the main characteristics of NB-hop genes.
These genes encode for proteins that are involved in metabolic response to hypoxia. Univariate analysis of overall survival in the batch-adjusted training set based on the NB-hop genes showed that high expression of PGK1, PDK1, MTFP1, and FAM162a genes was associated with a significantly higher risk of death (hazard ratio (HR) > 1 and
p-value < 0.05;
Table 2), while a high expression of ALDOC was associated with a lower risk of death (HR < 1 and
p-value < 0.05;
Table 2).
2.4. Generation and Validation of a NB-hop Classifier for Predicting NB Patient Prognosis
The classifier was built from the expression of NB-hop genes and patient outcome in the training set using the LibSVM library and the leave one-out cross validation (LOOCV) technique (
Section 4). The predictive power of the NB-hop classifier was then estimated in the test set. NB-hop classifier predicted 414 out of 550 patients (75%) at favorable prognosis (F) and 136 out of 550 patients (25%) at unfavorable prognosis (UF). Moreover, it was able to stratify patients into subgroups that had a significantly different overall survival (OS) and event-free survival (EFS) (OS: HR 5.2 95% confidence interval (CI) 7.3–16.3 and EFS: HR 3.3 95% CI 3.8–7.5, both
p < 0.0001;
Figure 2A).
Our classifier obtained a significant overall performance of 44% of Matthew’s correlation coefficient (MCC) (78% of Accuracy) in the test set (
Figure 2B and
Table S3). For comparison, we trained and tested four alternative machine learning algorithms on the batch-adjusted data set. The performance of each of these methods was lower than that of libSVM (MCC < 44%,
Table S4). To assess the significance of the NB-hop classifier performance, we applied the ConfusionMatrix function that was implemented in the Caret R Package [
29] to NB-hop prediction and event overall in the batch-adjusted test set. We found that the no information rate was 0.709 and the difference of accuracy between NB-hop classifier and no information rate was significant (
p value < 0.0001). These findings support our conclusion that the NB-hop classifier is an accurate predictor of NB patients’ outcome.
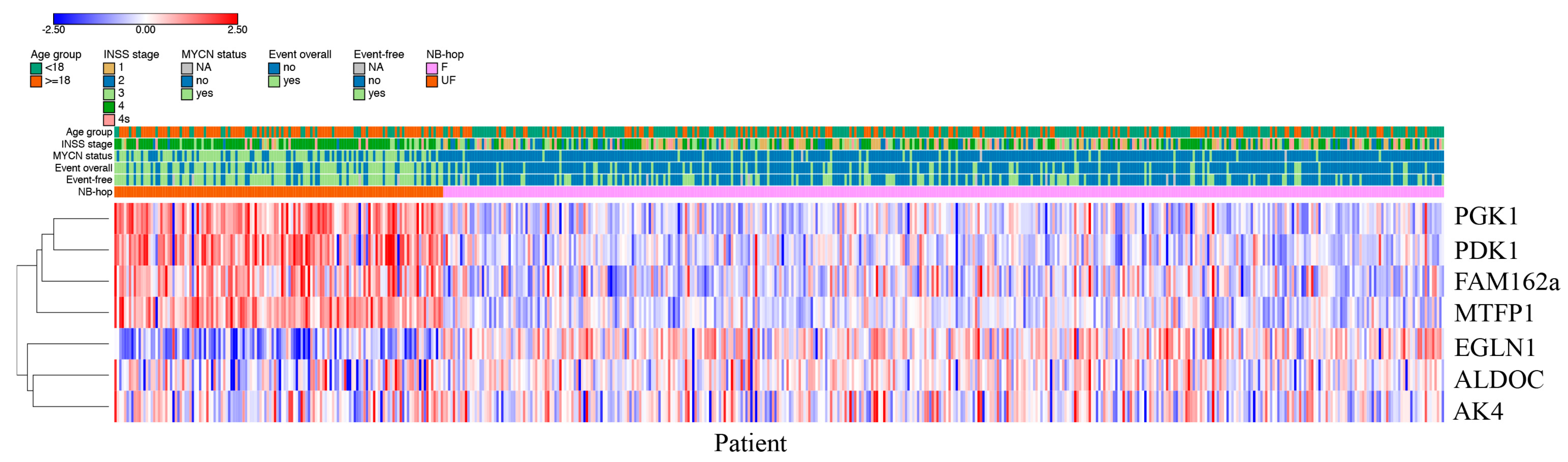
Patients predicted as UF by NB-hop had a clear different expression profile with respect to those predicted as F NB-hop, demonstrating that NB-hop was able to distinguish two groups of patients at the gene expression level (
Figure 3).
HIF-1a and HIF-2a are hypoxia inducible factor α-subunits that mediate the cellular response to hypoxia [
16]. We compared the distribution of HIF-1a and EPAS1/HIF-2a mRNA expression of NB patients grouped by NB-hop prediction in batch-adjusted test set. Box plots displayed in
Figure S3 show a significant up-regulation of HIF-1a and a significant down-regulation of EPAS1/HIF-2a expression in the group of UF NB-hop tumors (
p < 0.0001). These data indicate that UF NB-hop tumors are more hypoxic than F NB-hop tumors. We also correlated the HIF-1a gene expression and that of each NB-hop marker by Pearson correlation. We found a significant correlation between HIF1a and NB-hop markers (
p < 0.05).
The prognostic power of the NB-hop classifier was compared with that of established markers utilizing NB-hop classifier predictions (UF vs. F), INSS stage (4 vs. 1, 2, 3, 4 s), age at diagnosis (age ≥ 18 months vs. < 18 months), and MYCN status (amplified vs. single copy) in the test set. UF NB-hop, advanced stages 4, age ≥ 18 months, and MYCN amplification were significantly associated with a higher risk of death or undergoing an event according to a univariate analysis (HR > 1 and
p < 0.0001;
Table 3).
Importantly, the NB-hop classifier maintained a significant prognostic effect in the model adjusted for these clinical covariates in multivariate analysis (OS: HR 1.8 95% CI 1.2–2.6,
p = 0.004 and EFS: HR 1.7 95% CI 1.2–2.5,
p = 0.001;
Table 3). We concluded that NB-hop is an independent prognostic biomarker of unfavorable prognosis in NB.
We evaluated the distribution of the two populations of patients with F and UF prognosis in the subsets defined by age at diagnosis, INSS stage, MYCN status, and risk group. The number of patients predicted with UF NB-hop was greater than zero in all subsets, but it was higher in patients with age greater than 18 months, INSS stage 4, amplified MYCN, and high-risk disease (
Figure 4).
These results indicate that UF patients are associated with unfavorable clinical characteristics and suggest that NB-hop may additionally stratify clinical groups of patients. We assessed the stratification of sub-cohorts defined by known prognostic markers in order to test this hypothesis (
Table 4).
The NB-hop classifier significantly stratified patients with tumor stage 1, 2, 3, and 4, patients with age < 18 months, patients with age > 18 months, and patients with not amplified MYCN tumor (p < 0.05). The NB-hop classifier was not able to significantly stratify patients with amplified MYCN or stage 4s tumors (p > 0.05).
Next, the prognostic value of our predictor was assessed in additional clinically relevant subgroups of patients defined by combination of established prognostic markers. The group of high-risk patients older than 18 months with stage 4 tumors is a group traditionally difficult to stratify [
1]. NB-hop classifier identified two subgroups of patients with significantly different OS and EFS (OS: HR 1.8 95% CI 1.3–2.8,
p = 0.002 and EFS: HR 1.7 95% CI 1.2–2.5,
p = 0.008;
Figure 5A).
The NB-hop classifier achieved a significant prediction performance of 21% MCC (Fisher
p-value = 0.009). In the subgroup of low and intermediate-risk patients with stage 4 tumors lacking MYCN amplification and age < 18 months, NB-hop classifier identified two groups of patients with significant different EFS (EFS: HR 4.6 95% CI 2.5–69.5,
p = 0.006;
Figure 5B), but did not significantly discriminate patients who died from disease in this subgroup (OS: HR 4.1 95% CI 0.9–55.7,
p = 0.1;
Figure 5B).
In the subset of patients with stage 1, 2, 4s tumors lacking MYCN amplification, who are notoriously at low-risk of dying of disease [
2], our classifier was able to discriminate patients who underwent an event from those who did not (EFS: HR 5.1 95% CI 6.8–220.6,
p < 0.0001
Figure 5C). On the contrary, NB-hop was not able to significantly stratify patients who died, even though we observed a positive trend (OS:
p > 0.05,
Figure 5C). Patients that were older than 18 months with stage 3 and not amplified MYCN tumor constitute a group of localized tumors at intermediate risk, whose stratification remains a challenge [
30]. NB-hop classifier was able to additionally stratify this population of patients (OS: HR 3.9 95% CI 1.3–42.9 and EFS: HR 4.2 95% CI 1.5–52.4,
p ≤ 0.05
Figure 5D).
These findings highlight the ability of NB-hop to stratify NB patients that are difficult to stratify with actual risk assignment, indicating the potential prognostic significance of this classifier in NB.
2.5. Comparative Transcriptome Analysis of Primary Tumor Specimens between NB-hop Unfavorable and Favorable Patients
Microarray and RNA-seq platforms provide expression quantification of several transcripts in parallel from a sample. We compared the tumor transcription profile between F and UF populations of NB patients predicted by NB-hop classifier in the test set in order to investigate the key molecular mechanisms altered in NB hypoxic TME. Expression differences ≥1.5 or ≤−1.5 and Benjamini–Hochberg q-value ≤0.05 were considered to be statistically significant. Using these selection criteria, we identified 2377 differentially expressed genes (DEGs) (
Table S5). Among them, 926 were up-regulated and 1451 were down-regulated in UF NB-hop with respect to F NB-hop tumors.
Pathway analysis is a well-known bioinformatic tool that is used to explore the biological processes and pathways associated with a list of differentially expressed genes using curated ontologies [
31]. The list of DEGs was analyzed by the STRING-DB software using Gene ontology (GO), KEGG, and REACTOME ontologies (see Materials and Methods). Pathway analysis of up and down-regulated genes identified 546 and 705 significantly enriched biological processes and pathways in UF NB-hop, respectively.
Table S6 reports the complete list of significant processes and pathways for every analysis.
Table 5 presents a selection of significantly enriched functional terms.
Up-regulated genes were mainly involved in the cellular response to hypoxia, telomere maintenance, chromatin remodeling, DNA damage response, P53 mediated cell cycle arrest, cellular senescence, cell cycle, and proliferation. Down-regulated genes were mainly involved in immune response, cell differentiation, motility, inflammation response, cell death, and angiogenesis. These results showed that patients with UF NB-hop tumor are characterized by a hypoxic, immune suppressive, poorly differentiated, and apoptosis-resistant TME.
Table 6 lists DEGs whose role has been previously reported in NB.
The up- or down-regulation of these genes have been previously associated with an unfavorable prognosis of NB patients, in accordance with our results. Among the up-regulated genes, several were involved in glycolysis (GAPDH, HK2, PGK1, PKM, LDHA, LDHB, and SLCO4A1), pH regulation (SLC16A1), and homeostasis (PDK1), indicating a metabolic reprogramming typical of hypoxic cells. In addition, a prominent set of up-regulated genes coding for proteins with a primary role in telomere maintenance (FEN1, PCNA, and TERT), DNA damage response (BRCA1, BRCA2, CHEK1, CHEK2, and TPX2), P53 mediated cell cycle arrest (CDK1, CDK2, and TP53), and proliferation (AURKA, ERBB4, LIN28B, LMO3, MYCN, ODC1, and RAN). On the contrary, we demonstrated the down-regulation of genes coding for proteins that are involved in immune responses (CADM1, CCL19, CCL2, CCR7, CD226, and CXCL12), cell differentiation (CAMTA1, DNER, NTRK1, and PTN), cell motility and invasion (CD44, CD9, CDH1, ERBB3, L1CAM, NRP1, and SRCIN1), and angiogenesis (EPAS1). We also found the down-regulation of genes coding for proteins that are involved in chromatin remodeling (ARID1A and CHD5) and of the MYC gene and the up-regulation of genes involved in cell differentiation (ALK, PROM1, and RET), and coding for proteins that are involved in apoptosis and cell death (BIRC5 and TWIST1).
We performed a network analysis using STRING-DB software in order to assess the biological connection between HIF-1a and genes reported in
Table 6 [
89]. The resulting network that is shown in
Figure S4 displayed significantly more interactions than expected for a random set of proteins of similar size drawn from the genome (PPI enrichment
p-value < 1.0 × 10
−16). Our findings establish a connection between HIF-1a and genes reported in
Table 6 and indicate that those protein-coding genes are biologically connected as a group.
2.6. Correlating Immune Markers with NB-Hop Prediction and HIF1 Signature
The estimation of the abundance of immune and stromal cell populations in the TME may uncover their role in tumor development/progression [
90]. Differential expression analysis provides useful information regarding deregulated genes, but it is not suitable for estimating the abundance of different tumor-infiltrating cells. For this purpose, we applied the microenvironment cell populations (MCP)-counter method using the expression profile of NB tumors in the batch-adjusted test set. MCP-counter is a very well-known and widely used bioinformatic tool. Furthermore, transcriptome-based cell-type quantification methods for immuno-oncology are valuable tools with several successful applications [
91]. MCP-counter returned an abundance score for each cell type and tumor sample in the test set.
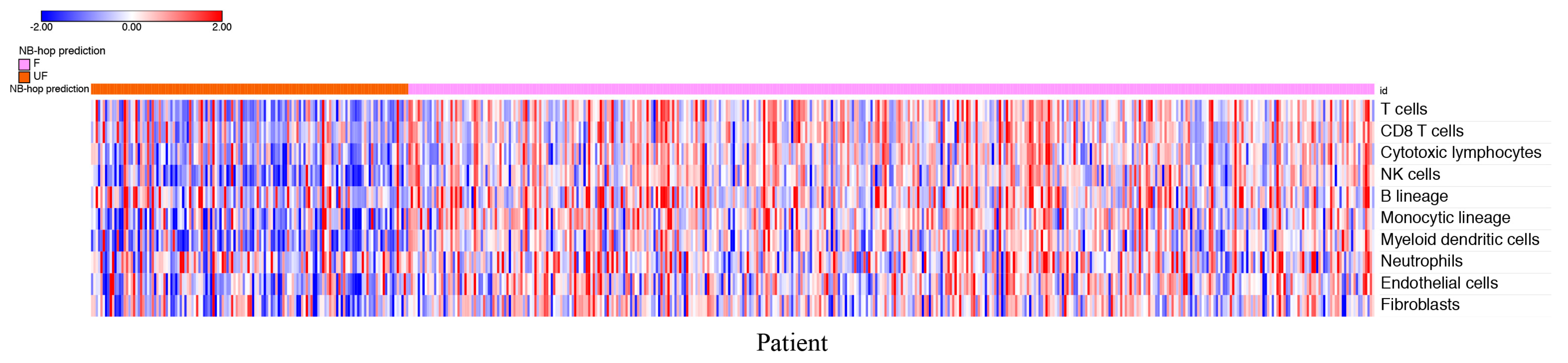
Figure 6 depicts the heat map of the scores for each patient grouped by NB-hop prediction.
The heat map shows a clear tendency to a lower abundance of T cells, CD8+ T cells, NK cells, cytotoxic lymphocytes, B cell lineage, monocytic lineage cells, myeloid dendritic cells, endothelial cells, and fibroblasts in patients with UF NB-hop tumors than in F NB-hop tumors, suggesting the association between an immunosuppressive TME and UF NB-hop tumors.
We carried out additional analyses to provide more links between MCP-counter analysis and hypoxia. For each cell type MCP-counter defines a set of characteristic genes that we refer to as MCP-counter markers [
90]. Overlap between MCP-counter markers and genes in the HIF-1a interaction network may provide relevant evidence of a direct involvement of HIF-1a in the regulation of immune or non-immune cell population activity. To this end, we assessed the overlap between the list of MCP-counter markers and the list of genes in the HIF-1a interaction network. BioGRID is a public curated gene interaction repository [
92]. Three-hundred and eighty-nine HIF-1a interactor genes were retrieved from the BioGRID repository. The resulting HIF-1a interaction network is shown in
Figure S5. Venn diagram showed that none of the MCP-counter markers were in the HIF-1a interaction network. Network analysis is a valuable tool for assessing the functional interaction among a set of genes. We performed a network analysis for HIF-1a and MCP-counter markers using the STRING-DB software [
89]. Interestingly, we found that HIF-1a was functionally associated with MCP-counter markers (
Figure S6). These results showed a significant functional association among HIF-1a and MCP-counter markers, raising the question of the possible correlation between HIF-1a and MCP-counter markers in NB. Thus, we carried out a correlation between the expression of HIF-1a and that of each MCP-counter marker in the batch-adjusted test set using Pearson’s correlation method. The results reported in
Table S7 showed that high HIF-1a expression is negatively correlated with most of the markers of T cells, CD8 T cells, cytotoxic lymphocytes, NK cells, myeloid dendritic cells cell types (Pearson r < 0 and
p value < 0.05,
Table S7), and positively correlated with most of the makers of monocytic lineage, neutrophils, endothelial cells and fibroblasts (Pearson r > 0 and
p value < 0.05,
Table S7). Within the set of significantly correlated immune markers in
Table S7, we found that MAL, BANK1, CXCR2, and KCNJ15 genes have been previously reported to play a tumor suppressive role in different types of cancer [
93,
94,
95,
96]. These findings support the results shown in
Figure 6.
The presence of specific immune regulatory cell populations is associated with poor outcome in different types of cancer, including NB [
97]. Tumor infiltrating macrophages (TAMs) often display an immunosuppressive M2-phenotype in aggressive NB tumors [
97]. Gene expression analysis of primary human NB tumors without MYCN amplification has revealed that high-level expression of TAM-specific genes, including CD14, CD16, IL6, IL6R, and TGFB1, was associated with poor five-year event-free survival [
98]. We assessed the correlation between the expression of HIF-1a and that of TAM-specific genes in the batch-adjusted test set with not amplified MYCN tumors in order to find additional associations between hypoxia and immune suppressive TME in NB. The results reported in
Table S8 showed a significant positive correlation between HIF-1a and CD14, IL6, and IL6R, while no significant correlation was found between HIF-1a and TGFB1. CD16/FCGR3A was absent in the batch-adjusted data set and it was excluded from the analysis. These findings suggest that hypoxia is associated with the presence of TAMs in NB without MYCN amplification supporting our conclusion that hypoxia is an unfavorable prognostic marker and is associated with an immune suppressive TME in NB.
One of the main mechanisms exploited by tumors for immune system escape is modulating immune checkpoints, inhibitory pathways that physiologically maintain self-tolerance, and limit the duration and amplitude of immune responses [
99]. We focused on the immune checkpoint ligands PD-L1 (CD274), PD-L2 (PDCD1LG2), and B7-H3 (CD276), which are recognized by inhibitory receptors that are expressed by T and NK cytolytic immune effectors [
99]. Because no PD-L2 gene expression was detected in the batch-adjusted data set, a comparison between UF and F NB-hop tumors was carried out for B7-H3 and PD-L1 expression. The results presented in
Figure S7 showed that B7-H3 was significantly up-regulated, whereas PD-L1 was significantly down-regulated in UF NB-hop tumors, thus indicating a differential mRNA expression modulation of these immune checkpoints in NB tumors.
2.7. Validation of the NB-hop Classifier for Predicting TERT Gene Over-expression in an Independent Cohort of NB Patients
We used the gene expression profiles of 262 untreated primary NB tumors (Agilent262), for the majority of which we had information on the TMM activation in addition to other patient clinical and molecular data, such as age at diagnosis, ALT activation, event overall, event-free, TERT rearrangement, INSS stage, MYCN status, p53/RAS gene mutations, and spontaneous regression, in order to evaluate the association between hypoxia and TMM in NB [
13]. Patients included in the data set did not overlap with the 786 patients in the batch-adjusted data set. Similarly to that described for the batch-adjusted data set, a classifier based on the expression of the 7 NB-hop genes and patient outcome was built in the training set (54 patients equal to 21%) using the LibSVM library and LOOCV technique. The predictive power of the NB-hop classifier was validated in the test set (208 patients equal to 79%). NB-hop classifier predicted a favorable prognosis for 138 out of 208 patients (66%) and an unfavorable prognosis for 70 out of 208 patients (33%). The clinical and molecular characteristics and NB-hop predictions of patients are reported in
Table S9.
We, then, analyzed the distribution of TERT mRNA expression in the profiles of patients belonging to the Agilent262 test set that was grouped by NB-hop predictions. We found that TERT mRNA expression was significantly elevated in patients with UF prognosis with respect to those predicted with F prognosis (
p < 0.00001,
Figure 7).
These findings confirmed the association between hypoxia and telomerase activity/expression, raising the question of the importance of hypoxia for TMM in NB.
2.8. Univariate Regression Analysis for Assessing the Association between Hypoxia and Telomere Maintenance Mechanisms in NB
We investigated the association between hypoxia, TMM, and other available clinical and molecular prognostic factors for NB [
13] in the Agilent262 test set by univariate logistic regression analysis. This analysis showed that patients with UF expression of the NB-hop biomarker had higher odds of having an age > 18 months, of undergoing an event or relapse/progression, INSS 4 stage tumor, MYCN amplification, p53/RAS gene mutations, or TMM (odd ratio > 1 and
p-value < 0.05,
Table 7). The association between NB-hop and TMM is still significant even excluding amplified MYCN tumors from the data set (Odd ratio > 1
p < 0.05).
On the contrary, patients with documented tumor regression were associated with a lower odd of UF NB-hop prognosis (odd ratio < 1 and
p-value < 0.05,
Table 7). No significant odd ratio was found between NB-hop prediction and ALT or TERT rearrangements, which indicated that NB-hop prediction is not related to these factors (
p-value > 0.05,
Table 7). Heat map visualization of NB-hop gene expression and established prognostic factors for NB showed that UF NB-hop patients had a clear different expression profile with respect to F NB-hop ones, confirming that NB-hop was able to distinguish two distinct groups of patients with different prognosis at the gene expression level (
Figure 8).
Furthermore, heat map graphically showed the association between the UF NB-hop subgroup and the worse clinical and molecular characteristic of NB patients, in accordance with the univariate regression analysis. These findings provide an indication that hypoxia is associated with telomerase activity, thus representing a potential critical factor for the classification and treatment of NB patients.
3. Discussion
Novel prognostic markers and therapeutic targets are urgently needed in the clinical settings to improve prognosis of NB patient and design more effective treatments and better-tailored therapies addressing NB heterogeneity [
1]. Here, we propose a new seven-gene hypoxia biomarker, referred to as NB-hop, as an unfavorable prognostic marker for NB patients.
The NB-hop biomarker was defined by gene expression analysis carried out on 1620 tumor specimens belonging to three available data sets, whose expression was measured by three different gene expression platforms, which included Agilent, Illumina, and Affymetrix technologies. The RNA-seq498, Agilent709, and Affymetrix413 data sets were homogeneous on the basis of patient clinical and molecular data and they were used for dissecting the prognostic value of the new NB-hop hypoxia biomarker. Analysis of the gene expression profile of NB primary tumors to find prognostic factors is an active research field, and several prognostic signatures have been reported [
5,
6,
100]. However, a large-scale expression study of NB tumors has not been previously carried out because different technologies used different proprietary annotations to identify transcripts. Meta-analysis is traditionally used in retrospective studies to overcome this problem [
28]. However, meta-analysis must be replicated independently in each data set, limiting the statistical power of the study [
28]. Here, we used a novel approach that is based on the application of the batch effect removal methods [
28] for integrating the RNA-seq498, Agilent709, and Affymetrix413 data sets [
28]. Data integration raises the question of the presence of the so-called batch effect [
28]. Our analysis evidenced that the simple integration of the NB datasets introduces a measurable batch effect, which could hinder subsequent analysis. Several batch effect removal methods have been described in the literature [
28]. In the present study, we used COMBAT, a well-known technique to remove batch effect for data integration [
28]. The application of this method to integrate gene expression data of NB tumor samples profiled by different platforms allowed us to build up one single multiplatform and multicenter cohort of 786 patients, with at least five years of follow-up, representing the largest data set described so far in a gene expression study of NB patients.
The expression profiles of 30% NB tumor samples served to build a classifier and the profiles of the remaining 70% were used to test its prognostic value in a validation data set. Split a data set into a training set and a test set is a standard machine learning procedure for computing an unbiased estimation of the performances of a classifier. In the test set, the NB-hop classifier distinguished two groups of patients at the gene expression level. The UF NB-hop group was composed by 25% of samples and was referred as the group with hypoxic tumor and unfavorable prognosis. F NB-hop was composed by 75% of samples and referred as the group with normoxic tumor and favorable prognosis. OS and EFS were significantly lower for UF NB-hop respect to F NB-hop patients. UF NB-hop tumors were associated with unfavorable clinical characteristics including age > 18 months, INSS stage 4, amplified MYCN, or high-risk disease, which confirmed the unfavorable prognosis of UF NB-hop patients. We demonstrated that NB-hop retains its independent prognostic effect in multivariable analysis that included age at diagnosis, INSS stage, and MYCN status, which are considered the strongest prognostic clinical and molecular variables for prediction of OS and EFS in NB [
2]. Analysis was focused on established risk factors in NB, such as age at diagnosis, MYCN status, and INSS stage, whose data were available in the original data sets, but not on other known risk factors, such as chromosomal aberration, ploidy, grade of differentiation, or histological category [
2], because these data were not reported in the original publications.
The clinical value of novel prognostic factors is often evaluated in selected groups of patients defined by combination of established NB risk factors [
5,
6,
100]. In this study, we assessed the prognostic value of our biomarker in clinically relevant groups of NB patients whose stratification was not previously reported. The groups included patients with metastatic (INSS stage 4) disease and age > 18 months, patients with age < 18 months, metastatic disease (INSS stage 4) and not amplified MYCN, and patients with localized (INSS stage 1, 2) or metastatic (INSS stage 4s) tumors with not amplified MYCN, and patients with age > 18 months, stage 3 and not amplified MYCN that are classified as high, intermediate, and low risk according to INRG schema [
1,
2,
30]. We demonstrated that NB-hop additionally stratified these groups of patients, making of NB-hop a new potentially eligible biomarkers for inclusion in upcoming NB pre-treatment risk schema.
NB-hop classifier is a valuable tool with potential direct application in the clinical management of children that are affected by NB. A classifier may be used to discriminate at diagnosis patients with hypoxic and unfavorable tumor from those with normoxic and favorable one. For patients with hypoxic and unfavorable tumor, classifier may be used for additionally refining patient risk stratification, optimizing treatment, and reducing unnecessary therapy related toxicity that remain among the most relevant open questions in the clinical discussion [
3,
4,
5,
6]. Nowadays, the inclusion of new prognostic biomarkers into the clinical practice is limited, probably because of the relatively small number of patients used so far in similar studies and the consequent limited reproducibility of the analyses. Our classifier is robust, because it was trained and tested using a large number of gene expression profiles measured with different platforms. Still, a potential limitation of the applicability of our classifier may depend on NB tumor samples that are available for gene expression profile necessary to determine the hypoxic status of these tumors. In fact, it is often difficult to surgically obtain tumor samples from young children.
NB-hop genes are known from the literature to be modulated by hypoxia and encode proteins that are involved in key biological processes associated with the response to hypoxia [
3,
17,
101,
102,
103,
104,
105,
106,
107,
108,
109]. Expression of these genes has been previously reported to represent a marker of hypoxia in NB cell lines [
3,
102,
105] and tumors [
102], and to correlate with NB bone marrow metastases and poor patient outcome [
3,
101]. The expression of NB-hop genes correlates with HIF-1a expression, which is one of the most important regulators of the cellular response to hypoxia [
16].
Low oxygen levels are known to induce remarkable transcriptional changes that affect the entire TME [
20]. Several scientific publications have evidenced the effects of NB cells adaptation to the hypoxic stress in terms of cell cycle arrest, cell differentiation, cellular stemness, resistance to cell death, genomic instability, cellular motility, invasiveness, and proliferation [
14,
23,
110]. However, the molecular mechanisms mediating the NB tumor response to the hypoxic TME in NB have been only partially understood [
23]. Our study is the first one assessing the hypoxia-deregulated transcription program in a large set of NB tumors.
A comparison of the expression profiles between UF and F NB-hop tumors showed the significant modulation of a large number of genes. Two distinct types of analysis were used in order to identify the pathways and cellular processes associated with the expression of these genes. Firstly, we utilized a pathway analysis based on three public ontologies. This approach was instrumental to identify the most affected biological processes and pathways associated with UF NB-hop tumors. Secondly, we performed a bibliographic search to identify the genes whose role in the pathogenesis and prognosis of NB had already been reported. Our findings evidence that the UF NB-hop tumors display up-regulation of genes that are known to be induced by hypoxia and to be involved in the glycolytic pathway, pH regulation, and homeostasis [
3,
5,
32,
33,
34,
35]. Importantly, among the up-regulated genes in UF NB-hop tumors, we also identified HIF-1a. An elevated HIF-1a expression fits with hypoxic, aggressively growing, and necrotic NB tumors, thus confirming that NB-hop is able to predict the hypoxic status of NB.
We found distinct evidences of the association between hypoxia and TMM in NB. The enrichment of various pathways and processes that are involved in telomere maintenance as well as the up-regulation of TERT gene was, in fact, observed in the group of UF NB-hop tumors. The aberrant expression of TERT has been described to be closely associated with tumorigenesis [
111], and the activation of telomerase through over-expression of TERT is thought to represent the most common pathway for cellular immortalization [
25]. The expression of the TERT gene has been shown to correlate with telomerase activity in experiments involving NB tumor tissues [
112] and to be a prognostic marker in various adult and pediatric tumors, including NB, where high levels of telomerase expression/activity were found to predict poor outcome [
113,
114]. Telomere lengthening to achieve immortalization is also obtained through ALT [
24,
115], a telomerase-independent mechanism that accounts for 10–15% of NB tumors that has been associated with unfavorable prognosis in NB [
24]. We did not find an association between hypoxia and ALT mechanism for telomerase lengthening, while our findings support the conclusion that hypoxia induces telomere maintenance in NB through activation of telomerase-dependent mechanisms.
Although telomerase regulation by hypoxia through the HIF-1α, the main regulator of gene transcription by hypoxia, has been described in both cancer and normal cells [
116,
117,
118,
119,
120], the association between hypoxia and telomerase-dependent telomere maintenance has never been described in NB tumors. Thus, here we show, for the first time, that hypoxia may drive a novel potential telomerase-dependent mechanism for telomere maintenance in NB.
More than 60% of NB tumors are metastatic, and secondary tumor sites can be found in bone, bone marrow, liver, lymph nodes, or, less commonly, in the skin, lung, or brain [
121]. Metastasis is the main cause of NB-related deaths, making the investigation of the processes involved in tumor metastasization mandatory [
122]. Cell adhesion is the process by which cells interact and attach to neighboring cells through specialized glycoproteins of the cell surface called cell adhesion molecules (CAMs) [
123]. The loss of cell adhesion allows malignant cells to detach and escape from the primary mass gaining a more motile and invasive phenotype [
124]. CAMs, such as cadherin, integrins, selectins, immunoglobulins, and CD44, are known to play a key role in each step of the metastasization process and are expressed in NB tumor cells [
125]. Hypoxia promotes invasion and metastasization in NB by regulating CAMs expression [
121]. Our data highlight a clear association between hypoxic tumors and metastatic disease (INSS stage 4), and provide evidences of the down-regulation of a large set of genes that are involved in cell adhesion within the group of UF NB-hop tumors. Furthermore, the loss of cell adhesion may also occur as a consequence of down-regulation of the CD44, CDH1, and L1CAM, which are well known CAMs in NB [
76,
78,
81].
An important enhancer of cancer invasion and metastasis is epithelial-to-mesenchymal transition (EMT), a process that allows epithelial cells to adopt a more mesenchymal state with stem-like properties [
123]. EMT involves expression modulation of several genes, including CHD1, which encodes E-cadherin, one of the most important markers of EMT in several types of cancer, including NB [
126]. Other genes that are implicated in EMT in NB are TWIST1 [
127,
128], PLK4 [
83], KRT19 [
79], and ERBB3 [
79]. In addition, the down-regulation of NRP1, CD9, and SRCIN1 encoding genes was reported to induce metastasis and invasion in NB [
77,
82,
84]. Although hypoxia induces EMT in different types of cancer by modulating the expression of specific marker genes [
129], the association between hypoxia and EMT in NB tumors has not been investigated. We found down-regulation of CDH1, CD9, SRCIN1, NRP1, KRT19, ERBB3 expression, and up-regulation of TWIST1 and PLK4 in UF NB-hop tumors. Furthermore, our data indicate the potential poor differentiation state of neurons associated with UF NB-hop tumors, as shown by the down-regulation of NTRK1, CAMTA1, DNER, and PTN, as well as the modulation of the known neuronal differentiation markers of NB RET, ALK, and PROM1 [
67,
68,
69,
70,
72,
74]. These data indicate that hypoxic NB tumors display gene expression modulation compatible with a mesenchymal state with stem-like properties and an increased invasiveness and metastasization potential.
Cellular proliferation is a hallmark of cancer [
130]. A complex network of transcription factors, kinases, and cell cycle regulators is exploited by NB cells to proliferate [
130]. Up to now, the mechanisms that mediate NB cell proliferation in response to hypoxia have not been fully elucidated [
23]. In this study, we report that UF NB-hop tumors display the expression of genes that are involved in cellular proliferation, cell cycle, mitotic cell cycle, and cell division, among which we report AURKA, ERBB4, LIN28B, LMO3, MYCN, ODC1, and RAN genes, whose overexpression has been previously documented to induce proliferation in NB cells [
50,
51,
52,
53,
54,
55,
56].
The abundance and composition of immune cells that infiltrate the TME is a critical determinant of tumor development, therapy efficacy, and clinical outcome in NB [
22,
97]. Cytotoxic T lymphocytes, B lymphocytes, and NK cells are responsible for the elimination of cancer cells [
131,
132,
133,
134]. Their massive infiltration in the TME is associated with a better clinical outcome of NB [
22,
135,
136]. Monocytic-lineage cells are a heterogenous population of immune cells and a key component of the innate defense mechanisms that display a dual influence on tumor progression, having the potential to activate immunosurveillance and exert anti-tumor responses, but becoming subverted by the tumor to support its progression, spread, and immune evasion [
137,
138]. Myeloid dendritic cells (DCs) are professional antigen-presenting cells that are central to the orchestration of innate and acquired immunity which were described in the TME of many cancer types, and their inactivation was reported as one of the main mechanisms by which tumors can escape from immune surveillance [
139,
140]. Hypoxia is one of the critical signals conditioning the balance between immune cell anti-/pro-tumoral functions and it has been extensively reported to contribute to immune resistance and tumor escape mechanisms favoring an immunosuppressive TME in different types of cancer [
141]. The mechanisms adopted by NB cells to evade immune response and grow uncontested have been extensively reviewed [
142,
143]. However, the association between hypoxia and immune suppression has never been investigated in NB tumors. The data reported in this study show, for the first time, the association between an immunosuppressive TME and hypoxic NB tumors. We demonstrated the down-regulation of a large number of genes involved in T cell activation, lymphocyte activation and immune response and the down-regulation of genes previously reported to induce immune suppression in NB, including CADM1, CCL19, CCL2, CCR7, CD226, and CXCL12 [
58,
59,
60,
63,
65,
66]. Furthermore, we found low abundance of infiltrating cytotoxic T and NK cells, DCs, and monocytic lineage cells in UF NB-hop tumors by MCP-counter analysis [
90], highlighting the presence of immunosuppressive conditions in the hypoxic TME favorable for NB tumor growth and metastatic spread. These findings are in accordance with previous evidences reporting a low level of infiltrated lymphocytes, monocytes, and macrophages in the population of high-risk amplified MYCN primary metastatic NB tumors [
97]. However, overlapping analysis between the HIF-1a gene network and MCP-counter markers did not show a direct interaction between HIF-1a and MCP-counter markers. Instead, network analysis based on public knowledge highlighted an indirect functional association between HIF-1a and MCP-counter markers. Transcriptional regulation by hypoxia is a complex process that involves several regulatory elements, including NF-kB, Ets-1, C/EBPα/β, AP-1, and Egr-1 transcription factors [
144]. Therefore, not every effect of hypoxia is mediated by HIFs [
144]. These considerations may potentially explain the lack of direct interaction between HIF-1a and MCP-counter markers.
We decided to carry out a correlation analysis between HIF-1a and each MCP-counter marker in order to investigate this association in NB. Although network, correlation and MCP-counter analyses are based on different methodological and statistical approaches, results obtained with these methods support the association between immune suppression and hypoxic TME in NB. Because the tumor suppressing/promoting role of several immune markers remains controversial to date, we also assessed the potential suppressive role of the immune markers significantly correlated with HIF-1a. Bibliographic search evidenced that MAL, BANK1, CXCR2, and KCNJ15 genes play a tumor suppressive role in different types of cancer, including colorectal cancer, B-cell lymphoma, and renal cell carcinoma [
93,
94,
95,
96]. However, we did not find any study reporting the tumor suppressive role of these genes in NB.
The high expression of TAM-specific genes was associated with poor five-year event-free survival in not amplified MYCN tumors [
98]. In order to find additional associations between hypoxia and immune suppressive TME in NB, we correlated the expression of HIF-1a and that of prognostic TAM-specific genes. The significant correlation between the expression of HIF-1a and TAMs-specific genes supported the conclusion that hypoxia is an unfavorable prognostic marker and the potential association between immune suppression and a hypoxic TME in NB. These results are in agreement with previous findings that showed the association between cancer macrophage infiltration and immune suppression and highlighted the regulatory role of HIF-1a in cancer-associated macrophages polarization and infiltration [
20].
Immune checkpoints are important pathways that are exploited by tumor cells to escape immune system response [
99]. We analyzed the expression of B7-H3 and PD-Ls, immune checkpoints known to limit both the NK and T cell-mediated cytolytic activity, in order to assess the link between hypoxia and immune checkpoint expression in NB tumors. B7-H3 is highly expressed by NB and it is considered an unfavorable prognostic factor [
99,
145]. Our findings reported the up-regulation of B7-H3 in UF NB-hop tumors indicating a potential B7-H3-mediated immune regulation in this group of tumors. PD-L1 is up-regulated in NB by immunostimulatory cytokines, such as IFN-g [
99], and the effect of its expression on patient survival is controversial [
146]. Our results support an inverse correlation between PD-L1 expression and NB patient prognosis.
Despite the therapeutic advances in the last years, a large number of NB patients still undergo a refractory disease [
147]. The mechanisms inducing resistance to therapy has gained great attention [
147]. Apoptosis deregulation, the presence of cancer stem cells, alterations of drug target, augmented capacity of DNA repair, faster drug effluence, and autophagy have been reported as the main factors mediating NB cell acquisition of drug resistance [
44]. However, little is known regarding the effects of hypoxia on these processes. Hypoxia has been associated with treatment resistance to a number of anticancer agents. Graham and Unger have documented the efficacy of twenty-two cancer drugs in human cell lines of non-pediatric cancers that were cultured in vitro under varying oxygen concentrations [
18]. Five out of the six therapeutic agents conventionally used in the treatment of high-risk NB patients are among the cancer drugs whose efficacy was shown to be reduced by hypoxia [
18]. Hypoxia promotes the NB chemoresistance to etoposide, vincristine, and cisplatin, which are three chemotherapeutic drugs used in the front-line treatment of high-risk NB patients [
148,
149]. Chemotherapeutic agents induce catastrophic DNA disruption with the consequent cancer cell death. However, DNA repair mechanisms are induced in cancer cells to overcome drugs-associated DNA damages [
21]. In this study, we provide multiple evidences that UF NB-hop tumors up-regulate genes that are involved in different DNA damage response mechanisms, such as DNA repair, double-strand break repair, base-excision repair, and mismatch repair. Concordantly, we report the up-regulation of genes, including BRCA1, BRCA2, CHEK1, CHEK2, and TPX2, which play a regulatory role in the DNA damage response in NB [
40,
41,
42,
43,
44].
p53 is a transcription factor that is involved in cellular defense against malignant transformation [
48]. Hypoxia is a potent activator of p53 in different types of cancers [
23]. However, conflicting evidences exist on the status of activation of p53 pathway signaling in NB cells [
48]. Our findings highlight the up-regulation of genes that are involved in the p53 signaling pathway, p53-mediated cell cycle arrest, and cellular senescence, as well as the up-regulation of genes, including CDK1, CDK2, and TP53, whose overexpression indicates the activation of cell cycle arrest in NB [
45,
46,
48].
Cell growth arrest promotes cellular resistance to apoptosis, which is associated with an unfavorable prognosis [
150]. Our findings indicate that UF NB-hop tumors, with respect to F NB-hop tumors, down-regulate genes that are involved in cell death and up-regulate genes, including BIRC5 and TWIST1, whose anti-apoptotic role in NB cells has been previously documented [
85,
86,
87].
Altogether, our data indicate that NB patients with hypoxic tumors are potentially more susceptible to acquire resistance to conventional chemotherapeutic drugs than patients with normoxic tumors. We believe that information derived from the assessment of the hypoxic status of the tumors may potentially be used for taking chemotherapeutic decisions in order to improve treatment response and reduce the side effects that are associated with aggressive therapies.
Hypoxia is a potential therapeutic target and may offer several therapeutic opportunities in cancer treatment [
18,
151]. Therapeutic approaches targeting tumor hypoxia with hypoxia-activated prodrugs and topotecan [
152] was shown to improve tumor response and prolong survival in NB xenograft models [
153]. We believe that the combination of therapeutic agents targeting cancer cells and the TME counteracting the effects of inadequate tumor oxygenation could provide more effective anti-tumor immunity. The success of these new potential therapeutic advances depends on the assessment of the hypoxic status of the tumor at diagnosis. To this end, NB-hop may be instrumental to predict those NB patients who have a hypoxic tumor and may benefit from anti-hypoxia treatments.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}