Romo1 Inhibition Induces TRAIL-Mediated Apoptosis in Colorectal Cancer

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Romo1 Inhibition Enhances TRAIL Sensitivity in CRC
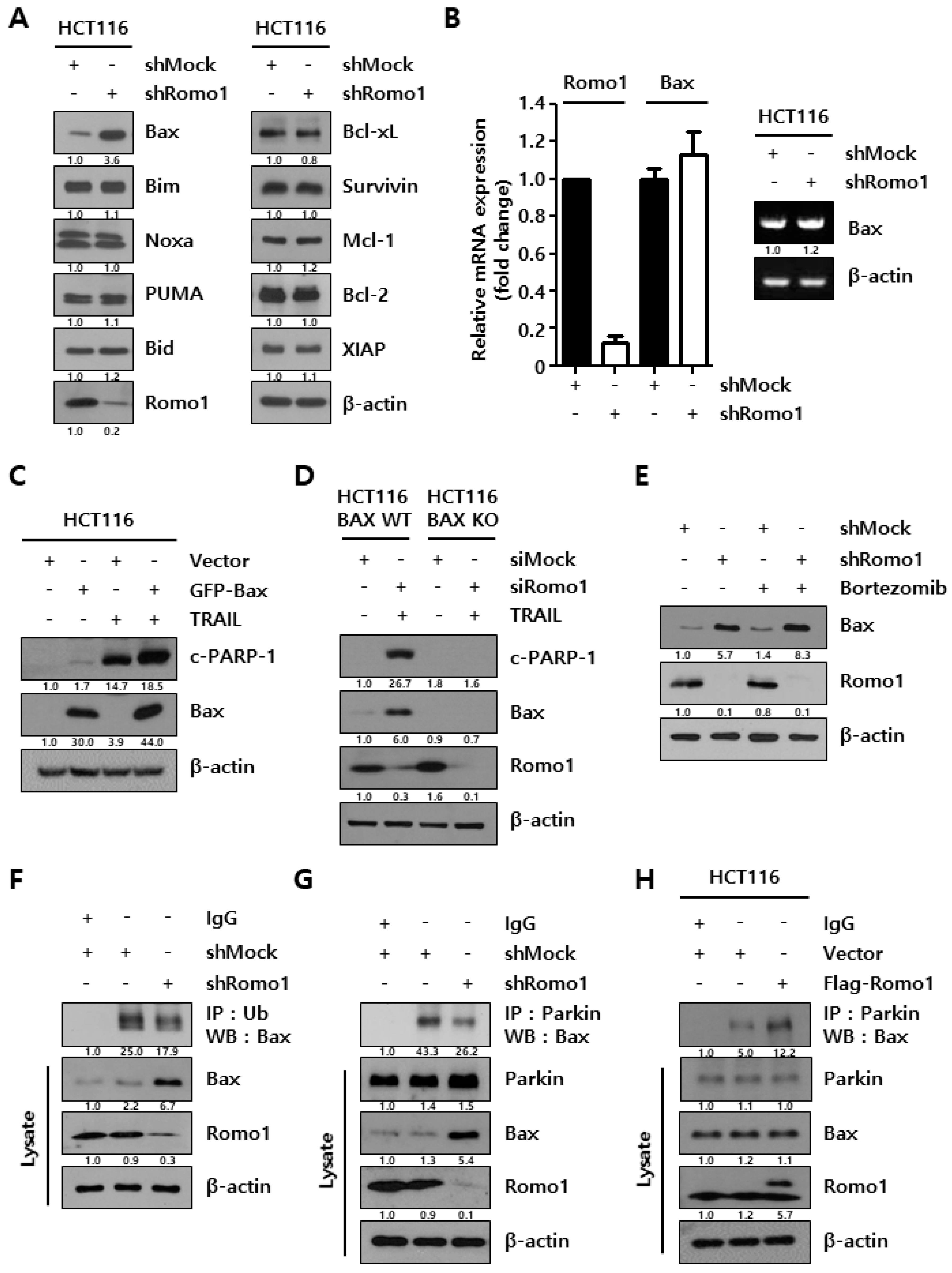
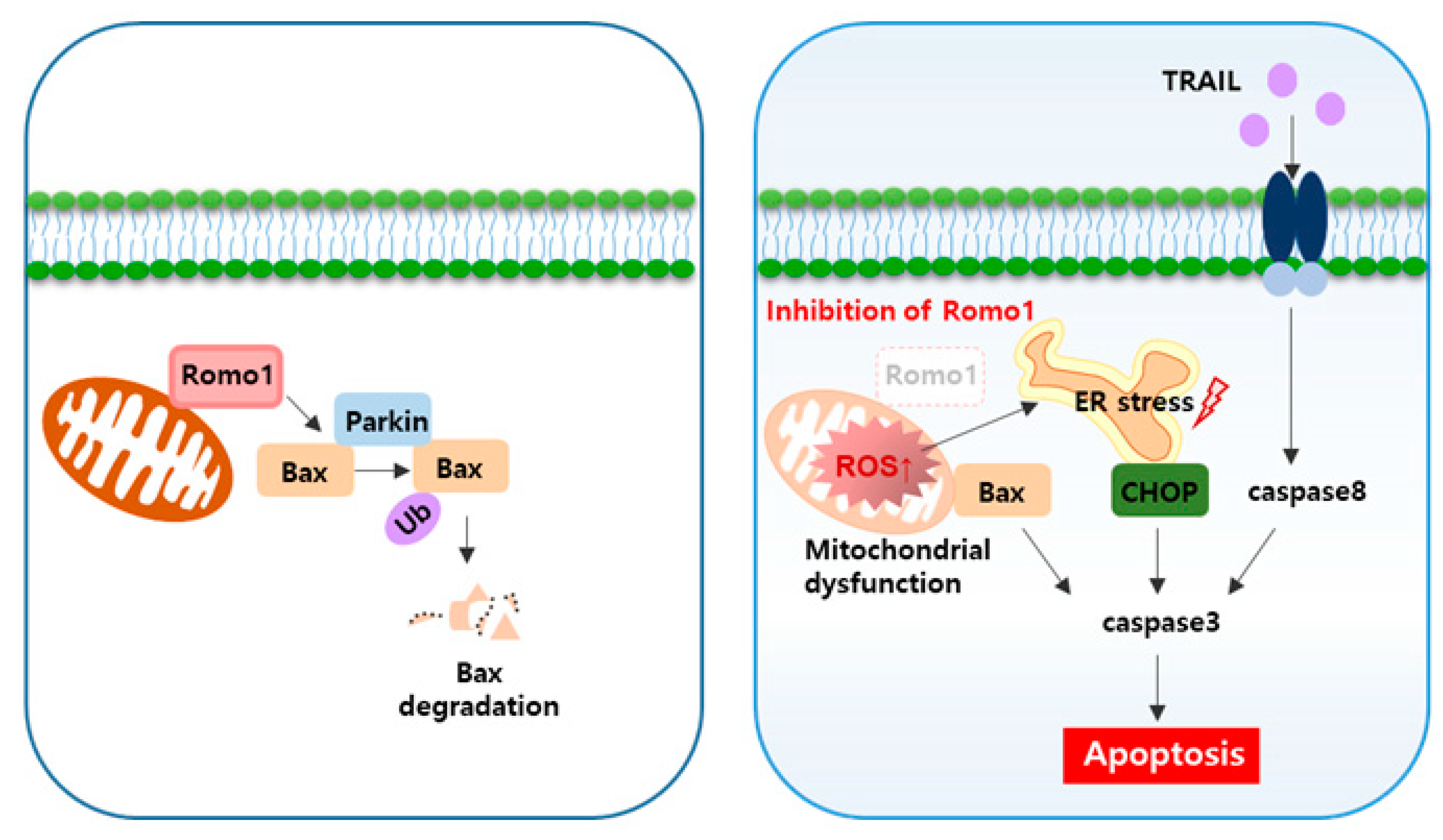
2.2. Romo1 Knockdown Decreases Bax Ubiquitination by Decreasing Interaction with Parkin
2.3. Romo1 Inhibition Triggers Mitochondrial Dysfunction and Loss of MMP
2.4. Mitochondrial Dysfunction upon Romo1 Inhibition Causes ROS
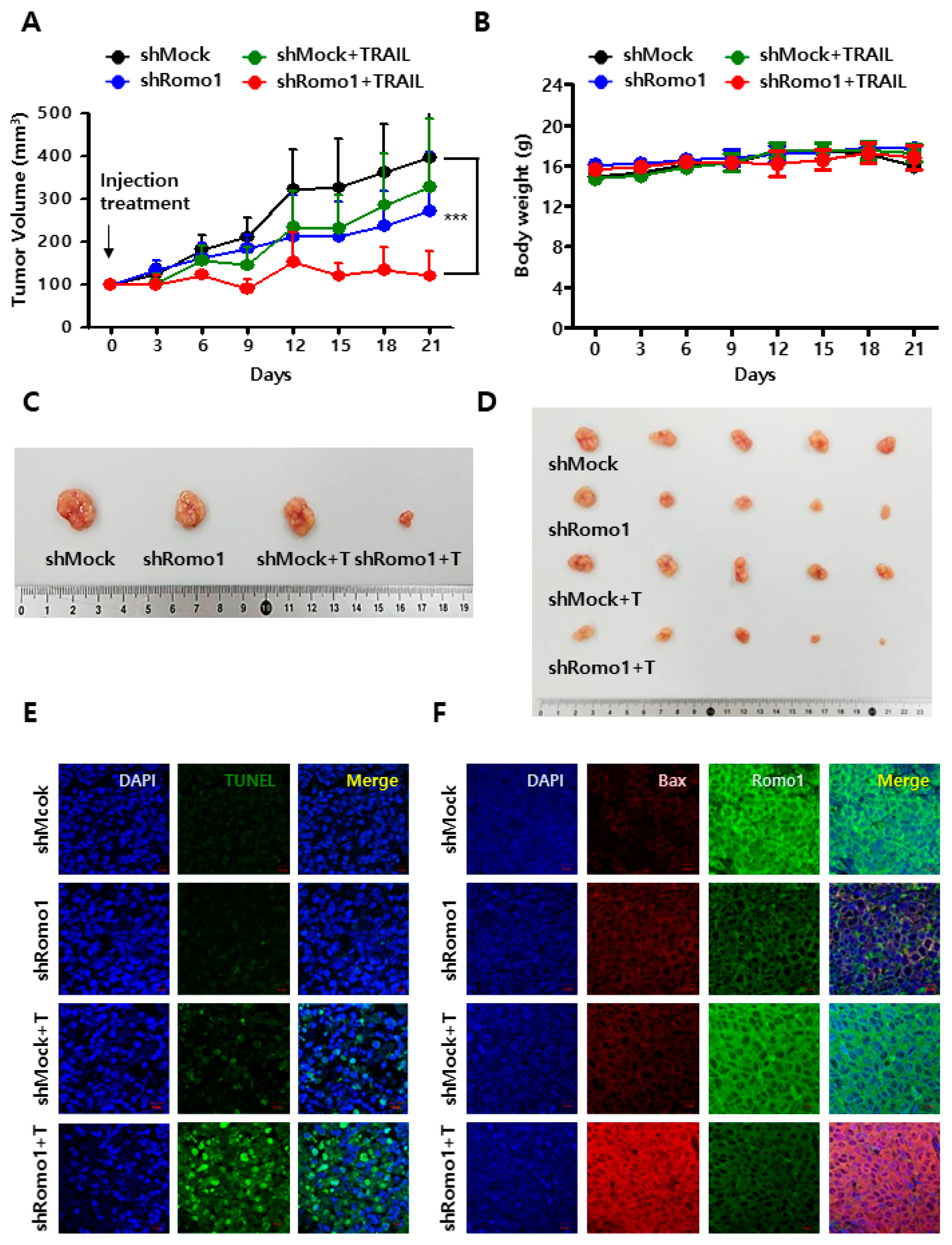
2.5. Romo1 Inhibition Promotes TRAIL-Induced Apoptosis In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transient and Stable Transfection
4.3. Reagents and Antibodies
4.4. Flow Cytometry Analysis
4.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.6. Quantitative Real Time PCR (qRT-PCR)
4.7. Immunoblotting
4.8. Co-Immunoprecipitation (Co-IP)
4.9. The Mitochondria and Cytosol Fractionation
4.10. Mitochondrial Membrane Potential (MMP) Measurement
4.11. Oxygen Consumption Rate (OCR)
4.12. Immunofluorescence Analysis
4.13. Immunohistochemical Analysis
4.14. TUNEL Assay
4.15. Tumor Xenograft In Vivo Model
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Paul, C.E.; Xu, X.N.; Boquan, J.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005, 15, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Mérino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: Present and future challenges. Expert Opin. Ther. Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuckey, D.W.; Shah, K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med 2013, 19, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jo, M.J.; Kim, B.R.; Kim, J.L.; Jeong, Y.A.; Na, Y.J.; Park, S.H.; Lee, S.Y.; Lee, D.H.; Lee, H.S.; et al. Reactive oxygen species modulator-1 (Romo1) predicts unfavorable prognosis in colorectal cancer patients. PLoS ONE 2017, 12, e0176834. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.M.; Kim, J.S.; Do Yoo, Y. A novel protein, Romo1, induces ROS production in the mitochondria. Biochem. Biophys. Res. Commun. 2006, 347, 649–655. [Google Scholar] [CrossRef]
- Chung, J.S.; Park, S.; Park, S.H.; Park, E.R.; Cha, P.H.; Kim, B.Y.; Chung, Y.M.; Woo, S.R.; Han, C.J.; Kim, S.B.; et al. Overexpression of Romo1 promotes production of reactive oxygen species and invasiveness of hepatic tumor cells. Gastroenterology 2012, 143, 1084–1094.e7. [Google Scholar] [CrossRef]
- Norton, M.; Ng, A.C.-H.; Baird, S.; Dumoulin, A.; Shutt, T.; Mah, N.; Andrade-Navarro, M.A.; McBride, H.M.; Screaton, R.A. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 2014, 7, ra10. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; You, D.-g.; Lee, H.-R.; Hwang, S.W.; Lee, C.J.; Yoo, Y.D. Romo1 is a mitochondrial nonselective cation channel with viroporin-like characteristics. J. Cell Biol. 2018, 217, 2059–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.N.; Berger, A.K.; Cortese, G.P.; Lavoie, M.J. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc. Natl. Acad. Sci. USA 2012, 109, 6283–6288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boohaker, R.J.; Zhang, G.; Carlson, A.L.; Nemec, K.N.; Khaled, A.R. BAX supports the mitochondrial network, promoting bioenergetics in nonapoptotic cells. Am. J. Physiol. Cell Physiol. 2011, 300, C1466–C1478. [Google Scholar] [CrossRef] [PubMed]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. CSH Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. Colorectal cancer chemotherapy: The evolution of treatment and new approaches. Curr. Med. Chem. 2017, 24, 1537–1557. [Google Scholar] [CrossRef]
- Mishra, J.; Drummond, J.; Quazi, S.H.; Karanki, S.S.; Shaw, J.J.; Chen, B.; Kumar, N. Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit. Rev. Oncol. Hemat. 2013, 86, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Köhne, C.H. Successes and limitations of targeted cancer therapy in colon cancer. Prog. Tumor Res. 2014, 41, 36–50. [Google Scholar] [PubMed]
- Arnold, D.; Prager, G.W.; Quintela, A.; Stein, A.; Moreno Vera, S.; Mounedji, N.; Taieb, J. Beyond second-line therapy in patients with metastatic colorectal cancer: A systematic review. Ann. Oncol. 2018, 29, 835–856. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.-h.; Wang, H.-y.; Li, J.; Huang, Y.-z. TRAIL-based gene delivery and therapeutic strategies. Acta Pharmacol. Sin. 2019, 40, 1373–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, S.; Chen, Q.; Sarva, K.; Siddiqui, I.; Srivastava, R.K. Curcumin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells: Molecular mechanisms of apoptosis, migration and angiogenesis. J. Mol. Signal. 2007, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Refaat, A.; Abdelhamed, S.; Yagita, H.; Inoue, H.; Yokoyama, S.; Hayakawa, Y.; Saiki, I. Berberine enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast cancer. Oncol. Lett. 2013, 6, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Refaat, A.; Abd-Rabou, A.; Reda, A. TRAIL combinations: The new ‘trail’ for cancer therapy (Review). Oncol. Lett. 2014, 7, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, Y.H.; Chung, J.S.; Yoo, Y.D. Romo1 and the NF-kappaB pathway are involved in oxidative stress-induced tumor cell invasion. Int. J. Oncol. 2015, 46, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Schwabe, R.F.; Qian, T.; Lemasters, J.J.; Brenner, D.A. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002, 36, 1498–1508. [Google Scholar]
- Keane, M.M.; Rubinstein, Y.; Cuello, M.; Ettenberg, S.A.; Banerjee, P.; Nau, M.M.; Lipkowitz, S. Inhibition of NF-kappaB activity enhances TRAIL mediated apoptosis in breast cancer cell lines. Breast Cancer Res. Treat. 2000, 64, 211–219. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, H.S.; Kim, S.U.; Noh, E.J.; Lee, J.S.; Choi, K.S. Sodium butyrate sensitizes human glioma cells to TRAIL-mediated apoptosis through inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene 2005, 24, 6877–6889. [Google Scholar] [CrossRef] [Green Version]
- Kurbanov, B.M.; Fecker, L.F.; Geilen, C.C.; Sterry, W.; Eberle, J. Resistance of melanoma cells to TRAIL does not result from upregulation of antiapoptotic proteins by NF-kappaB but is related to downregulation of initiator caspases and DR4. Oncogene 2007, 26, 3364–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, D.C.; Bakowska, J.C. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J. Vis. Exp. 2011, 51, 2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J. Natl Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Na, A.R.; Chung, Y.M.; Lee, S.B.; Park, S.H.; Lee, M.-S.; Do Yoo, Y. A critical role for Romo1-derived ROS in cell proliferation. Biochem. Biophys. Res. Commun. 2008, 369, 672–678. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Oikonomou, E.; Pintzas, A. The TRAIL of oncogenes to apoptosis. Biofactors 2013, 39, 343–354. [Google Scholar] [CrossRef]
- Rogler, G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014, 345, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, M. Ulcerative colitis-associated colorectal cancer. World J. Gastroenterol. 2014, 20, 16389–16397. [Google Scholar] [CrossRef]
- Lin, L.; Sun, Y.; Wang, D.; Zheng, S.; Zhang, J.; Zheng, C. Celastrol Ameliorates Ulcerative Colitis-Related Colorectal Cancer in Mice via Suppressing Inflammatory Responses and Epithelial-Mesenchymal Transition. Front. Pharmacol. 2016, 6, 320. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, M.J.; Kim, B.G.; Park, S.H.; Kim, H.J.; Jeong, S.; Kim, B.R.; Kim, J.L.; Na, Y.J.; Jeong, Y.A.; Yun, H.K.; et al. Romo1 Inhibition Induces TRAIL-Mediated Apoptosis in Colorectal Cancer. Cancers 2020, 12, 2358. https://doi.org/10.3390/cancers12092358
Jo MJ, Kim BG, Park SH, Kim HJ, Jeong S, Kim BR, Kim JL, Na YJ, Jeong YA, Yun HK, et al. Romo1 Inhibition Induces TRAIL-Mediated Apoptosis in Colorectal Cancer. Cancers. 2020; 12(9):2358. https://doi.org/10.3390/cancers12092358
Chicago/Turabian StyleJo, Min Jee, Bu Gyeom Kim, Seong Hye Park, Hong Jun Kim, Soyeon Jeong, Bo Ram Kim, Jung Lim Kim, Yoo Jin Na, Yoon A. Jeong, Hye Kyeong Yun, and et al. 2020. "Romo1 Inhibition Induces TRAIL-Mediated Apoptosis in Colorectal Cancer" Cancers 12, no. 9: 2358. https://doi.org/10.3390/cancers12092358