Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives


Abstract
:1. Introduction
2. Cutaneous Melanoma
2.1. Introduction
2.2. Genetic Landscape and Specific Treatment Approaches
2.2.1. NRAS
2.2.2. NF1
2.2.3. Uncommon BRAF Mutations
2.2.4. Actionable Mutations in the Triple Wild-Type Subgroup (No Mutations in BRAF, RAS, or NF1)
2.3. Entities with Special Morphology
2.3.1. Amelanotic Melanoma
2.3.2. Desmoplastic Melanoma
2.3.3. Spitzoid Melanoma
2.3.4. Acral Lentiginous Melanoma
3. Mucosal Melanoma
3.1. Introduction
3.2. Genetic Landscape and Targeted Therapy Approaches
3.2.1. BRAF
3.2.2. KIT
3.2.3. Others
3.3. Immunotherapy
4. Uveal Melanoma
4.1. Introduction
4.2. Genetic Landscape and Targeted Therapy Approaches
4.2.1. Gαq Signaling
4.2.2. Others
4.3. Tumor Immunogenicity and Therapy
5. Unusual or Unknown Primary Site Melanomas
5.1. Brain and Meninges
5.2. Primary Dermal Melanoma
5.3. Esophageal Melanoma
5.4. Primary Malignant Melanoma of the Breast and other UPSM
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thoelke, A.; Willrodt, S.; Hauschild, A.; Schadendorf, D. Primary extracutaneous malignant melanoma: A comprehensive review with emphasis on treatment. Onkologie 2004, 27, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, C.C.; Wu, X.-C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Pires da Silva, I.; Wang, K.Y.X.; Wilmott, J.S.; Holst, J.; Carlino, M.S.; Park, J.J.; Quek, C.; Wongchenko, M.; Yan, Y.; Mann, G.; et al. Distinct Molecular Profiles and Immunotherapy Treatment Outcomes of V600E and V600K BRAF-Mutant Melanoma. Clin. Cancer Res. 2019, 25, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Melis, C.; Rogiers, A.; Bechter, O.; van den Oord, J.J. Molecular genetic and immunotherapeutic targets in metastatic melanoma. Virchows Arch. Int. J. Pathol. 2017, 471, 281–293. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Lond. Engl. 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: A systemic review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.-Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Lebbe, C.; Dutriaux, C.; Lesimple, T.; Kruit, W.; Kerger, J.; Thomas, L.; Guillot, B.; de Braud, F.; Garbe, C.; Grob, J.J.; et al. Pimasertib (PIM) versus dacarbazine (DTIC) in patients (pts) with cutaneous NRAS melanoma: A controlled, open-label phase II trial with crossover. Ann. Oncol. 2016, 27, vi389. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Bastholt, L.; Robert, C.; Sosman, J.; Larkin, J.; Hersey, P.; Middleton, M.; Cantarini, M.; Zazulina, V.; Kemsley, K.; et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 2012, 18, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.W.; Lee, J.; Shin, S.J.; Kim, J.-S.; Kim, Y.J.; Han, H.S.; Lee, S.J.; Lim, H.-S.; Hong, Y.; Noh, Y.S.; et al. Belvarafenib, a novel pan-RAF inhibitor, in solid tumor patients harboring BRAF, KRAS, or NRAS mutations: Phase I study. J. Clin. Oncol. 2019, 37, 3000. [Google Scholar] [CrossRef]
- Schuler, M.H.; Ascierto, P.A.; De Vos, F.Y.F.L.; Postow, M.A.; Van Herpen, C.M.L.; Carlino, M.S.; Sosman, J.A.; Berking, C.; Long, G.V.; Weise, A.; et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J. Clin. Oncol. 2017, 35, 9519. [Google Scholar] [CrossRef]
- Algazi, A.P.; Esteve-Puig, R.; Nosrati, A.; Hinds, B.; Hobbs-Muthukumar, A.; Nandoskar, P.; Ortiz-Urda, S.; Chapman, P.B.; Daud, A. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 2018, 31, 110–114. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, M.C.; Ugurel, S.; Mangana, J.; Heppt, M.V.; Eigentler, T.K.; Berking, C.; Schadendorf, D.; Schuler, G.; Dummer, R.; Heinzerling, L. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 2018, 98, 10–16. [Google Scholar] [CrossRef]
- Johnson, D.B.; Lovly, C.M.; Flavin, M.; Panageas, K.S.; Ayers, G.D.; Zhao, Z.; Iams, W.T.; Colgan, M.; DeNoble, S.; Terry, C.R.; et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol. Res. 2015, 3, 288–295. [Google Scholar] [CrossRef] [Green Version]
- A Phase Ia/Ib Clinical Study to Evaluate the Safety, Pharmacokinetics (PK) and Preliminary Anti-Tumor Activity of FCN-159 in Patients with Advanced Melanoma Harboring NRAS-Aberrant (Ia) and NRAS-Mutant (Ib). Available online: https://clinicaltrials.gov/ct2/show/NCT03932253 (accessed on 25 June 2020).
- ClinicalTrials.gov, MEK and Autophagy Inhibition in Metastatic/Locally Advanced, Unresectable Neuroblastoma RAS (NRAS) Melanoma (CHLOROTRAMMEL). Available online: https://clinicaltrials.gov/ct2/show/NCT03979651 (accessed on 25 June 2020).
- ClinicalTrials.gov, Nivolumab and Trametinib with or without Dabrafenib in Treating Patients with BRAF Mutated or Wild Type Metastatic Stage III-IV Melanoma That Cannot Be Removed by Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT02910700 (accessed on 25 June 2020).
- Shahabi, V.; Whitney, G.; Hamid, O.; Schmidt, H.; Chasalow, S.D.; Alaparthy, S.; Jackson, J.R. Assessment of association between BRAF-V600E mutation status in melanomas and clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 733–737. [Google Scholar] [CrossRef]
- Dinter, L.; Karitzky, P.; Kaeubler, T.; Niessner, H.; Wanke, I.; Beissert, S.; Meier, F.; Westphal, D. BRAF/MEK inhibitors induce potent ER stress—Enforced apoptosis in BRAFwt/NRASmut melanoma cells—Insights into mode of action and resistance mechanisms. In Proceedings of the 16th International Society for Melanoma Research Congress, Salt Lake City, UT, USA, 20–21 November 2019; p. 249. [Google Scholar]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 Mutations Are Common in Desmoplastic Melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garman, B.; Anastopoulos, I.N.; Krepler, C.; Brafford, P.; Sproesser, K.; Jiang, Y.; Wubbenhorst, B.; Amaravadi, R.; Bennett, J.; Beqiri, M.; et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017, 21, 1936–1952. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Kong, B.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkhasyan, K.A.; Rooney, S.L.; Snow, A.N.; Swick, B.L.; Milhem, M.M.; Zakharia, Y. The clinical characteristics of melanoma with BRAF V600R mutation: A case series study. Melanoma Res. 2020, 30, 107–112. [Google Scholar] [CrossRef]
- Klein, O.; Clements, A.; Menzies, A.M.; O’Toole, S.; Kefford, R.F.; Long, G.V. BRAF inhibitor activity in V600R metastatic melanoma. Eur. J. Cancer 2013, 49, 1073–1079. [Google Scholar] [CrossRef]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.M.; Johnson, D.B.; Meiss, F.; et al. Targeted Therapy in Advanced Melanoma With Rare BRAF Mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef] [Green Version]
- Lokhandwala, P.M.; Tseng, L.-H.; Rodriguez, E.; Zheng, G.; Pallavajjalla, A.; Gocke, C.D.; Eshleman, J.R.; Lin, M.-T. Clinical mutational profiling and categorization of BRAF mutations in melanomas using next generation sequencing. BMC Cancer 2019, 19, 665. [Google Scholar] [CrossRef]
- Coit, D.G.; Thompson, J.A.; Algazi, A.; Andtbacka, R.; Bichakjian, C.K.; Carson, W.E.; Daniels, G.A.; DiMaio, D.; Fields, R.C.; Fleming, M.D. NCCN guidelines insights: Melanoma, version 3.2020. J. Natl. Compr. Cancer Netw. 2020, 14, 945–958. [Google Scholar]
- Busam, K.J.; Vilain, R.E.; Lum, T.; Busam, J.A.; Hollmann, T.J.; Saw, R.P.M.; Coit, D.C.; Scolyer, R.A.; Wiesner, T. Primary and Metastatic Cutaneous Melanomas Express ALK Through Alternative Transcriptional Initiation. Am. J. Surg. Pathol. 2016, 40, 786–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezcano, C.; Shoushtari, A.N.; Ariyan, C.; Hollmann, T.J.; Busam, K.J. Primary and Metastatic Melanoma With NTRK Fusions. Am. J. Surg. Pathol. 2018, 42, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.-T.; Zhou, Q.-M.; Wang, F.; Shao, Q.; Guan, Y.-X.; Wen, X.-Z.; Chen, L.-Z.; Feng, Q.-S.; Li, W.; Zeng, Y.-X.; et al. Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment Cell Melanoma Res. 2013, 26, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Couts, K.; Sheren, J.; Saichaemchan, S.; Ariyawutyakorn, W.; Avolio, I.; Cabral, E.; Glogowska, M.; Amato, C.; Robinson, S.; et al. Kinase gene fusions in defined subsets of melanoma. Pigment Cell Melanoma Res. 2017, 30, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handolias, D.; Salemi, R.; Murray, W.; Tan, A.; Liu, W.; Viros, A.; Dobrovic, A.; Kelly, J.; McArthur, G.A. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 2010, 23, 210–215. [Google Scholar] [CrossRef]
- Tzen, C.-Y.; Wu, Y.-H.; Tzen, C.-Y. Characterization of KIT mutation in melanoma. Dermatol. Sin. 2014, 32, 7–12. [Google Scholar] [CrossRef]
- Pizzichetta, M.A.; Talamini, R.; Stanganelli, I.; Puddu, P.; Bono, R.; Argenziano, G.; Veronesi, A.; Trevisan, G.; Rabinovitz, H.; Soyer, H.P. Amelanotic/hypomelanotic melanoma: Clinical and dermoscopic features. Br. J. Dermatol. 2004, 150, 1117–1124. [Google Scholar] [CrossRef]
- Thomas, N.E.; Kricker, A.; Waxweiler, W.T.; Dillon, P.M.; Busman, K.J.; From, L.; Groben, P.A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; et al. Comparison of clinicopathologic features and survival of histopathologically amelanotic and pigmented melanomas: A population-based study. JAMA Dermatol. 2014, 150, 1306–1314. [Google Scholar] [CrossRef]
- Cheung, W.L.; Patel, R.R.; Leonard, A.; Firoz, B.; Meehan, S.A. Amelanotic melanoma: A detailed morphologic analysis with clinicopathologic correlation of 75 cases. J. Cutan. Pathol. 2012, 39, 33–39. [Google Scholar] [CrossRef]
- Zalaudek, I.; Meiklejohn, W.; Argenziano, G.; Thurber, A.E.; Sturm, R.A. “White” nevi and “red” melanomas: Association with the RHC phenotype of the MC1R gene. J. Investig. Dermatol. 2009, 129, 1305–1307. [Google Scholar] [CrossRef]
- Ghiorzo, P.; Pastorino, L.; Pizzichetta, M.A.; Bono, R.; Queirolo, P.; Talamini, R.; Annessi, G.; Bruno, W.; Nasti, S.; Gargiulo, S.; et al. CDKN2A and MC1R analysis in amelanotic and pigmented melanoma. Melanoma Res. 2009, 19, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Moreau, J.F.; Weissfeld, J.L.; Ferris, L.K. Characteristics and survival of patients with invasive amelanotic melanoma in the USA. Melanoma Res. 2013, 23, 408–413. [Google Scholar] [CrossRef] [PubMed]
- McClain, S.E.; Mayo, K.B.; Shada, A.L.; Smolkin, M.E.; Patterson, J.W.; Slingluff, C.L. Amelanotic melanomas presenting as red skin lesions: A diagnostic challenge with potentially lethal consequences. Int. J. Dermatol. 2012, 51, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Pouryazdanparast, P.; Brenner, A.; Haghighat, Z.; Guitart, J.; Rademaker, A.; Gerami, P. The role of 8q24 copy number gains and c-MYC expression in amelanotic cutaneous melanoma. Mod. Pathol. 2012, 25, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Detrixhe, A.; Libon, F.; Mansuy, M.; Nikkels-Tassoudji, N.; Rorive, A.; Arrese, J.E.; Quatresooz, P.; Reginster, M.-A.; Nikkels, A.F. Melanoma masquerading as nonmelanocytic lesions. Melanoma Res. 2016, 26, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Beyeler, M.; Dummer, R. Cutaneous melanoma: Uncommon presentations. Clin. Dermatol. 2005, 23, 587–592. [Google Scholar] [CrossRef]
- Vernali, S.; Waxweiler, W.T.; Dillon, P.M.; Kanetsky, P.A.; Orlow, I.; Luo, L.; Busam, K.J.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; et al. Association of Incident Amelanotic Melanoma With Phenotypic Characteristics, MC1R Status, and Prior Amelanotic Melanoma. JAMA Dermatol. 2017, 153, 1026–1031. [Google Scholar] [CrossRef]
- Sturm, R.A.; Fox, C.; McClenahan, P.; Jagirdar, K.; Ibarrola-Villava, M.; Banan, P.; Abbott, N.C.; Ribas, G.; Gabrielli, B.; Duffy, D.L.; et al. Phenotypic characterization of nevus and tumor patterns in MITF E318K mutation carrier melanoma patients. J. Investig. Dermatol. 2014, 134, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Massi, D.; Pinzani, P.; Simi, L.; Salvianti, F.; De Giorgi, V.; Pizzichetta, M.A.; Mirri, F.; Steffan, A.; Orlando, C.; Santucci, M.; et al. BRAF and KIT somatic mutations are present in amelanotic melanoma. Melanoma Res. 2013, 23, 414–419. [Google Scholar] [CrossRef]
- Palla, B.; Su, A.; Binder, S.; Dry, S. SOX10 expression distinguishes desmoplastic melanoma from its histologic mimics. Am. J. Dermatopathol. 2013, 35, 576–581. [Google Scholar] [CrossRef]
- Chen, L.L.; Jaimes, N.; Barker, C.A.; Busam, K.J.; Marghoob, A.A. Desmoplastic melanoma: A review. J. Am. Acad. Dermatol. 2013, 68, 825–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, W.G.; Busam, K.J.; Ben-Porat, L.; Panageas, K.S.; Coit, D.G.; Gyorki, D.E.; Linehan, D.C.; Brady, M.S. Desmoplastic melanoma: A pathologically and clinically distinct form of cutaneous melanoma. Ann. Surg. Oncol. 2005, 12, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Boussemart, L.; Johnson, A.; Schrock, A.B.; Pal, S.K.; Frampton, G.M.; Fabrizio, D.; Chalmers, Z.; Lotem, M.; Gibney, G.; Russell, J.; et al. Tumor mutational burden and response to programmed cell death protein 1 inhibitors in a case series of patients with metastatic desmoplastic melanoma. J. Am. Acad. Dermatol. 2019, 80, 1780–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhill, R.L. The Spitzoid lesion: Rethinking Spitz tumors, atypical variants, “Spitzoid melanoma” and risk assessment. Mod. Pathol. 2006, 19 (Suppl. 2), S21–S33. [Google Scholar] [CrossRef] [Green Version]
- Lallas, A.; Kyrgidis, A.; Ferrara, G.; Kittler, H.; Apalla, Z.; Castagnetti, F.; Longo, C.; Moscarella, E.; Piana, S.; Zalaudek, I.; et al. Atypical Spitz tumours and sentinel lymph node biopsy: A systematic review. Lancet Oncol. 2014, 15, e178–e183. [Google Scholar] [CrossRef]
- Ghazi, B.; Carlson, G.W.; Murray, D.R.; Gow, K.W.; Page, A.; Durham, M.; Kooby, D.A.; Parker, D.; Rapkin, L.; Lawson, D.H.; et al. Utility of lymph node assessment for atypical spitzoid melanocytic neoplasms. Ann. Surg. Oncol. 2010, 17, 2471–2475. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [Green Version]
- Lazova, R.; Pornputtapong, N.; Halaban, R.; Bosenberg, M.; Bai, Y.; Chai, H.; Krauthammer, M. Spitz nevi and Spitzoid melanomas: Exome sequencing and comparison with conventional melanocytic nevi and melanomas. Mod. Pathol. 2017, 30, 640–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, S.S.; Peternel, S.; Mully, T.W.; North, J.P.; Pincus, L.B.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod. Pathol. 2020, 33, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.; Nagarajan, P.; Curry, J.L.; Tang, Z.; Kim, T.-B.; Aung, P.P.; Torres-Cabala, C.A.; Eterovic, A.K.; Wargo, J.A.; Prieto, V.G.; et al. Spitzoid melanoma with histopathological features of ALK gene rearrangement exhibiting ALK copy number gain: A novel mechanism of ALK activation in spitzoid neoplasia. Br. J. Dermatol. 2019, 180, 404–408. [Google Scholar] [CrossRef]
- Research, A.A. Potentially Actionable MAP3K8 Alterations Are Common in Spitzoid Melanoma. Cancer Discov. 2019, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Hanna, G.J.; Scholz, C.R.; Gualberto, A.; Park, S.H. Preliminary activity of tipifarnib in tumors of the head and neck, salivary gland and urothelial tract with HRAS mutations. J. Clin. Oncol. 2020, 38, 6504. [Google Scholar] [CrossRef]
- Bellew, S.; Del Rosso, J.Q.; Kim, G.K. Skin cancer in asians: Part 2: Melanoma. J. Clin. Aesthetic Dermatol. 2009, 2, 34–36. [Google Scholar]
- Dika, E.; Veronesi, G.; Altimari, A.; Riefolo, M.; Ravaioli, G.M.; Piraccini, B.M.; Lambertini, M.; Campione, E.; Gruppioni, E.; Fiorentino, M.; et al. BRAF, KIT, and NRAS Mutations of Acral Melanoma in White Patients. Am. J. Clin. Pathol. 2020, 153, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.R.; Choi, Y.D.; Kim, J.M.; Jin, S.; Shin, M.-H.; Shim, H.-J.; Lee, J.-B.; Yun, S.J. Genetic Alterations in Primary Acral Melanoma and Acral Melanocytic Nevus in Korea: Common Mutated Genes Show Distinct Cytomorphological Features. J. Investig. Dermatol. 2018, 138, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, S.E.; Davies, M.A. Targeting KIT in melanoma: A paradigm of molecular medicine and targeted therapeutics. Biochem. Pharmacol. 2010, 80, 568–574. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Xu, T.; Dai, J.; Ma, M.; Tang, H.; Chi, Z.; Si, L.; Cui, C.; Sheng, X.; Kong, Y.; et al. TERT copy gain predicts the outcome of high-dose interferon α-2b therapy in acral melanoma. OncoTargets Ther. 2018, 11, 4097–4104. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Yu, J.; Wu, X.; Guo, Q.; Yin, T.; Cheng, Z.; Dai, J.; Kong, Y.; Guo, J. The TERT copy number gain is sensitive to telomerase inhibitors in human melanoma. Clin. Sci. 2020, 134, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Eton, O.; Davis, D.W.; Frazier, M.L.; McConkey, D.J.; Diwan, A.H.; Papadopoulos, N.E.; Bedikian, A.Y.; Camacho, L.H.; Ross, M.I.; et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br. J. Cancer 2008, 99, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.-A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Kim, K.M.; Kwon, M.; Kim, J.H.; Lee, J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Investig. New Drugs 2012, 30, 2008–2014. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, T.M.; Kim, Y.J.; Jang, K.-T.; Lee, H.J.; Lee, S.N.; Ahn, M.S.; Hwang, I.G.; Lee, S.; Lee, M.-H.; et al. Phase II Trial of Nilotinib in Patients With Metastatic Malignant Melanoma Harboring KIT Gene Aberration: A Multicenter Trial of Korean Cancer Study Group (UN10-06). Oncologist 2015, 20, 1312–1319. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef]
- Delyon, J.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.-P.; Avril, M.-F.; Bedane, C.; Bens, G.; et al. STAT3 Mediates Nilotinib Response in KIT-Altered Melanoma: A Phase II Multicenter Trial of the French Skin Cancer Network. J. Investig. Dermatol. 2018, 138, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Kalinsky, K.; Lee, S.; Rubin, K.M.; Lawrence, D.P.; Iafrarte, A.J.; Borger, D.R.; Margolin, K.A.; Leitao, M.M.; Tarhini, A.A.; Koon, H.B.; et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 2017, 123, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.R.; Kashani-Sabet, M.; Garrido, M.; O’Day, S.J.; Hamid, O.; Bastian, B.C. Sunitinib therapy for melanoma patients with KIT mutations. Clin. Cancer Res. 2012, 18, 1457–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbie, R.; Ferguson, P.; Molina-Aguilar, C.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019, 247, 539–551. [Google Scholar] [CrossRef]
- Johnson, D.B.; Peng, C.; Abramson, R.G.; Ye, F.; Zhao, S.; Wolchok, J.D.; Sosman, J.A.; Carvajal, R.D.; Ariyan, C.E. Clinical Activity of Ipilimumab in Acral Melanoma: A Retrospective Review. Oncologist 2015, 20, 648–652. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef] [Green Version]
- Tacastacas, J.D.; Bray, J.; Cohen, Y.K.; Arbesman, J.; Kim, J.; Koon, H.B.; Honda, K.; Cooper, K.D.; Gerstenblith, M.R. Update on primary mucosal melanoma. J. Am. Acad. Dermatol. 2014, 71, 366–375. [Google Scholar] [CrossRef]
- Patrick, R.J.; Fenske, N.A.; Messina, J.L. Primary mucosal melanoma. J. Am. Acad. Dermatol. 2007, 56, 828–834. [Google Scholar] [CrossRef]
- Kuk, D.; Shoushtari, A.N.; Barker, C.A.; Panageas, K.S.; Munhoz, R.R.; Momtaz, P.; Ariyan, C.E.; Brady, M.S.; Coit, D.G.; Bogatch, K.; et al. Prognosis of Mucosal, Uveal, Acral, Nonacral Cutaneous, and Unknown Primary Melanoma From the Time of First Metastasis. Oncologist 2016, 21, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Furney, S.J.; Turajlic, S.; Stamp, G.; Nohadani, M.; Carlisle, A.; Thomas, J.M.; Hayes, A.; Strauss, D.; Gore, M.; van den Oord, J.; et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J. Pathol. 2013, 230, 261–269. [Google Scholar] [CrossRef]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef] [PubMed]
- DeMatos, P.; Tyler, D.S.; Seigler, H.F. Malignant melanoma of the mucous membranes: A review of 119 cases. Ann. Surg. Oncol. 1998, 5, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Spencer, S.A.; Lydiatt, W. Mucosal melanoma: A clinically and biologically unique disease entity. J. Natl. Compr. Cancer Netw. 2012, 10, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer International Publishing, Ltd.: Chicago, IL, USA, 2017; pp. 163–167. [Google Scholar]
- Kelly, P.; Zagars, G.K.; Cormier, J.N.; Ross, M.I.; Guadagnolo, B.A. Sphincter-sparing local excision and hypofractionated radiation therapy for anorectal melanoma: A 20-year experience. Cancer 2011, 117, 4747–4755. [Google Scholar] [CrossRef]
- Lian, B.; Si, L.; Cui, C.; Chi, Z.; Sheng, X.; Mao, L.; Li, S.; Kong, Y.; Tang, B.; Guo, J. Phase II randomized trial comparing high-dose IFN-α2b with temozolomide plus cisplatin as systemic adjuvant therapy for resected mucosal melanoma. Clin. Cancer Res. 2013, 19, 4488–4498. [Google Scholar] [CrossRef] [Green Version]
- Lian, B.; Guo, J. Adjuvant therapy of mucosal melanoma. Chin. Clin. Oncol. 2014, 3, 33. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Dumaz, N.; Jouenne, F.; Delyon, J.; Mourah, S.; Bensussan, A.; Lebbé, C. Atypical BRAF and NRAS Mutations in Mucosal Melanoma. Cancers 2019, 11, 1133. [Google Scholar] [CrossRef] [Green Version]
- Bryce, A.H.; Arguello, D.; Millis, S.Z.; Bender, R.; Reddy, S.K.; Gatalica, Z.; Gonzalez, R. Multiplatform biomarker analysis on non-sun exposed mucosal melanoma. J. Clin. Oncol. 2015, 33, 9042. [Google Scholar] [CrossRef] [Green Version]
- Nassar, K.W.; Tan, A.C. The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 2020, 61, 139–148. [Google Scholar] [CrossRef]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Bai, X.; Mao, L.L.; Chi, Z.H.; Sheng, X.N.; Cui, C.L.; Kong, Y.; Dai, J.; Wang, X.; Li, S.M.; Tang, B.X.; et al. BRAF inhibitors: Efficacious and tolerable in BRAF-mutant acral and mucosal melanoma. Neoplasma 2017, 64, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.-W.; Kim, S.-M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Rönnstrand, L. Signal transduction via the stem cell factor receptor/c-Kit. Cell. Mol. Life Sci. 2004, 61, 2535–2548. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F.; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Kong, Y.; Chi, Z.; Sheng, X.; Cui, C.; Wang, X.; Mao, L.; Tang, B.; Li, S.; Lian, B.; et al. MAPK Pathway and TERT Promoter Gene Mutation Pattern and Its Prognostic Value in Melanoma Patients: A Retrospective Study of 2,793 Cases. Clin. Cancer Res. 2017, 23, 6120–6127. [Google Scholar] [CrossRef] [Green Version]
- Cinotti, E.; Chevallier, J.; Labeille, B.; Cambazard, F.; Thomas, L.; Balme, B.; Leccia, M.T.; D’Incan, M.; Vercherin, P.; Douchet, C.; et al. Mucosal melanoma: Clinical, histological and c-kit gene mutational profile of 86 French cases. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1834–1840. [Google Scholar] [CrossRef]
- Toscano de Mendonça, U.B.; Cernea, C.R.; Matos, L.L.; Monteiro de Araujo Lima, R.R. Analysis of KIT gene mutations in patients with melanoma of the head and neck mucosa: A retrospective clinical report. Oncotarget 2018, 9, 22886–22894. [Google Scholar] [CrossRef] [Green Version]
- Omholt, K.; Grafström, E.; Kanter-Lewensohn, L.; Hansson, J.; Ragnarsson-Olding, B.K. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin. Cancer Res. 2011, 17, 3933–3942. [Google Scholar] [CrossRef] [Green Version]
- Carlino, M.S.; Todd, J.R.; Rizos, H. Resistance to c-Kit inhibitors in melanoma: Insights for future therapies. Oncoscience 2014, 1, 423–426. [Google Scholar] [CrossRef]
- Ablain, J.; Xu, M.; Rothschild, H.; Jordan, R.C.; Mito, J.K.; Daniels, B.H.; Bell, C.F.; Joseph, N.M.; Wu, H.; Bastian, B.C.; et al. Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 2018, 362, 1055–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Song, Z.; Chen, J.; Shepard, M.J.; Song, H.; Ren, G.; Li, Z.; Guo, W.; Zhuang, Z.; Shi, Y. Whole-exome sequencing of oral mucosal melanoma reveals mutational profile and therapeutic targets. J. Pathol. 2018, 244, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Shi, C.; Tao, W.; Li, J.; Wu, J.; Han, Y.; Yang, G.; Gu, Z.; Xu, S.; Wang, Y.; et al. Analysis of Mucosal Melanoma Whole-Genome Landscapes Reveals Clinically Relevant Genomic Aberrations. Clin. Cancer Res. 2019, 25, 3548–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Kong, Y.; Li, Y.; Zhang, Q.; Si, L.; Cui, C.; Chi, Z.; Tang, B.; Mao, L.; Lian, B.; et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur. J. Cancer 2016, 65, 156–163. [Google Scholar] [CrossRef]
- Lyu, J.; Wu, Y.; Li, C.; Wang, R.; Song, H.; Ren, G.; Guo, W. Mutation scanning of BRAF, NRAS, KIT, and GNAQ/GNA11 in oral mucosal melanoma: A study of 57 cases. J. Oral Pathol. Med. 2016, 45, 295–301. [Google Scholar] [CrossRef]
- Sari, Ş.Ö.; Yilmaz, İ.; Taşkin, O.Ç.; Narli, G.; Şen, F.; Çomoğlu, Ş.; Firat, P.; Bİlgİç, B.; Yilmazbayhan, D.; Özlük , Y.; et al. BRAF, NRAS, KIT, TERT, GNAQ/GNA11 mutation profile analysis of head and neck mucosal melanomas: A study of 42 cases. Pathology 2017, 49, 55–61. [Google Scholar] [CrossRef]
- Cosgarea, I.; Ugurel, S.; Sucker, A.; Livingstone, E.; Zimmer, L.; Ziemer, M.; Utikal, J.; Mohr, P.; Pfeiffer, C.; Pföhler, C.; et al. Targeted next generation sequencing of mucosal melanomas identifies frequent NF1 and RAS mutations. Oncotarget 2017, 8, 40683–40692. [Google Scholar] [CrossRef]
- Quek, C.; Rawson, R.V.; Ferguson, P.M.; Shang, P.; Silva, I.; Saw, R.P.M.; Shannon, K.; Thompson, J.F.; Hayward, N.K.; Long, G.V.; et al. Recurrent hotspot SF3B1 mutations at codon 625 in vulvovaginal mucosal melanoma identified in a study of 27 Australian mucosal melanomas. Oncotarget 2019, 10, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.D.; Holtan, S.G.; Ingle, J.N.; Croghan, G.A.; Kottschade, L.A.; Creagan, E.T.; Kaur, J.S.; Pitot, H.C.; Markovic, S.N. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer 2006, 106, 375–382. [Google Scholar] [CrossRef]
- Hodi, F.S.; Soiffer, R.J.; Clark, J.; Finkelstein, D.M.; Haluska, F.G. Phase II study of paclitaxel and carboplatin for malignant melanoma. Am. J. Clin. Oncol. 2002, 25, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Grob, J.J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A.; et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 2000, 18, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Mignard, C.; Deschamps Huvier, A.; Gillibert, A.; Duval Modeste, A.B.; Dutriaux, C.; Khammari, A.; Avril, M.-F.; Kramkimel, N.; Mortier, L.; Marcant, P.; et al. Efficacy of Immunotherapy in Patients with Metastatic Mucosal or Uveal Melanoma. J. Oncol. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Robert, C.; Ribas, A.; Hodi, F.S.; Walpole, E.; Daud, A.; Arance, A.S.; Brown, E.; Hoeller, C.; Mortier, L.; et al. Antitumour activity of pembrolizumab in advanced mucosal melanoma: A post-hoc analysis of KEYNOTE-001, 002, 006. Br. J. Cancer 2018, 119, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Moya-Plana, A.; Herrera Gómez, R.G.; Rossoni, C.; Dercle, L.; Ammari, S.; Girault, I.; Roy, S.; Scoazec, J.-Y.; Vagner, S.; Janot, F.; et al. Evaluation of the efficacy of immunotherapy for non-resectable mucosal melanoma. Cancer Immunol. Immunother. 2019, 68, 1171–1178. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35, 226–235. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Wagstaff, J.; Ascierto, P.A.; Butler, M.O.; Lao, C.D.; Marquez-Rodas, I.; Chiarion-Sileni, V.; Dummer, R.; Ferrucci, P.F.; Lorigan, P.; et al. CheckMate 067: Long-term outcomes in patients with mucosal melanoma. J. Clin. Oncol. 2020, 38, 10019. [Google Scholar] [CrossRef]
- Johnson, D.B.; Carlson, J.A.; Elvin, J.A.; Vergilio, J.-A.; Suh, J.; Ramkissoon, S.; Daniel, S.; Fabrizio, D.; Frampton, G.M.; Ali, S.M.; et al. Landscape of genomic alterations (GA) and tumor mutational burden (TMB) in different metastatic melanoma (MM) subtypes. J. Clin. Oncol. 2017, 35, 9536. [Google Scholar] [CrossRef]
- Johnson, B.F.; Clay, T.M.; Hobeika, A.C.; Lyerly, H.K.; Morse, M.A. Vascular endothelial growth factor and immunosuppression in cancer: Current knowledge and potential for new therapy. Expert Opin. Biol. Ther. 2007, 7, 449–460. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; LIAN, B.; Wang, X.; et al. Overall survival and biomarker analysis of a phase Ib combination study of toripalimab, a humanized IgG4 mAb against programmed death-1 (PD-1) with axitinib in patients with metastatic mucosal melanoma. J. Clin. Oncol. 2020, 38, 10007. [Google Scholar] [CrossRef]
- Si, L.; Sheng, X.; Mao, L.; Li, C.; Wang, X.; Bai, X.; Qi, Z.H.; Chi, Z.; Cui, C.; Lian, B.; et al. A phase II study of vorolanib (CM082) in combination with toripalimab (JS001) in patients with advanced mucosal melanoma. J. Clin. Oncol. 2020, 38, 10040. [Google Scholar] [CrossRef]
- Postow, M.A.; Luke, J.J.; Bluth, M.J.; Ramaiya, N.; Panageas, K.S.; Lawrence, D.P.; Ibrahim, N.; Flaherty, K.T.; Sullivan, R.J.; Ott, P.A.; et al. Ipilimumab for patients with advanced mucosal melanoma. Oncologist 2013, 18, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Del Vecchio, M.; Di Guardo, L.; Ascierto, P.A.; Grimaldi, A.M.; Sileni, V.C.; Pigozzo, J.; Ferraresi, V.; Nuzzo, C.; Rinaldi, G.; Testori, A.; et al. Efficacy and safety of ipilimumab 3mg/kg in patients with pretreated, metastatic, mucosal melanoma. Eur. J. Cancer 2014, 50, 121–127. [Google Scholar] [CrossRef]
- Si, L.; Zhang, X.; Shu, Y.; Pan, H.; Wu, D.; Liu, J.; Lou, F.; Mao, L.; Wang, X.; Wen, X.; et al. A Phase Ib Study of Pembrolizumab as Second-Line Therapy for Chinese Patients with Advanced or Metastatic Melanoma (KEYNOTE-151). Transl. Oncol. 2019, 12, 828–835. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Shah, S.U.; Luo, W.; Furuta, M.; Shields, J.A. Iris melanoma: Features and prognosis in 317 children and adults. J. Am. Assoc. Pediatric Ophthalmol. Strabismus 2012, 16, 10–16. [Google Scholar] [CrossRef]
- Koevary, S. Uveal Melanoma: An Updated Review. Biomed. Res. Rev. 2019, 2, 110. [Google Scholar]
- Gallagher, R.P.; Elwood, J.M.; Rootman, J.; Spinelli, J.J.; Hill, G.B.; Threlfall, W.J.; Birdsell, J.M. Risk factors for ocular melanoma: Western Canada Melanoma Study. J. Natl. Cancer Inst. 1985, 74, 775–778. [Google Scholar]
- Sliney, D.H. How light reaches the eye and its components. Int. J. Toxicol. 2002, 21, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Damato, E.M.; Damato, B.E. Detection and time to treatment of uveal melanoma in the United Kingdom: An evaluation of 2,384 patients. Ophthalmology 2012, 119, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Wetherill, C.; Cheong, J.; Jones, L.; Marshall, E.; Damato, B.; Coupland, S.E.; Ghaneh, P.; Poston, G.J.; Malik, H.Z.; et al. The Liverpool uveal melanoma liver metastases pathway: Outcome following liver resection. J. Surg. Oncol. 2014, 109, 542–547. [Google Scholar] [CrossRef]
- Rodrigues, M.; de Koning, L.; Coupland, S.E.; Jochemsen, A.G.; Marais, R.; Stern, M.-H.; Valente, A.; Barnhill, R.; Cassoux, N.; Evans, A.; et al. So Close, yet so Far: Discrepancies between Uveal and Other Melanomas. A Position Paper from UM Cure 2020. Cancers 2019, 11, 1032. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W. The genetics of uveal melanoma: An emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012, 25, 171–181. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Fitch, K.R.; Fuchs, H.; de Angelis, M.H.; Barsh, G.S. Effects of G-protein mutations on skin color. Nat. Genet. 2004, 36, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, F.; Caltabiano, G.; Queirolo, P. Uveal melanoma. Cancer Treat. Rev. 2012, 38, 549–553. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Luke, J.J.; Callahan, M.K.; Postow, M.A.; Romano, E.; Ramaiya, N.; Bluth, M.; Giobbie-Hurder, A.; Lawrence, D.P.; Ibrahim, N.; Ott, P.A.; et al. Clinical activity of ipilimumab for metastatic uveal melanoma: A retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial Sloan-Kettering Cancer Center, and University Hospital of Lausanne experience. Cancer 2013, 119, 3687–3695. [Google Scholar] [CrossRef]
- Piulats Rodriguez, J.M.; Ochoa de Olza, M.; Codes, M.; Lopez-Martin, J.A.; Berrocal, A.; García, M.; Gurpide, A.; Homet, B.; Martin-Algarra, S. Phase II study evaluating ipilimumab as a single agent in the first-line treatment of adult patients (Pts) with metastatic uveal melanoma (MUM): The GEM-1 trial. J. Clin. Oncol. 2014, 32, 9033. [Google Scholar] [CrossRef]
- Zimmer, L.; Vaubel, J.; Mohr, P.; Hauschild, A.; Utikal, J.; Simon, J.; Garbe, C.; Herbst, R.; Enk, A.; Kämpgen, E.; et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naïve patients with metastatic uveal melanoma. PLoS ONE 2015, 10, e0118564. [Google Scholar] [CrossRef]
- Joshua, A.M.; Monzon, J.G.; Mihalcioiu, C.; Hogg, D.; Smylie, M.; Cheng, T. A phase 2 study of tremelimumab in patients with advanced uveal melanoma. Melanoma Res. 2015, 25, 342–347. [Google Scholar] [CrossRef]
- Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.; Sosman, J.A.; et al. Efficacy and safety of programmed death receptor-1 (PD-1) blockade in metastatic uveal melanoma (UM). J. Clin. Oncol. 2016, 34, 9507. [Google Scholar] [CrossRef]
- Piulats, J.M.; De La Cruz-Merino, L.; Curiel Garcia, M.T.; Berrocal, A.; Alonso-Carrión, L.; Espinosa, E.; López Castro, R.; Rodriguez-Abreu, D.; Luna Fra, P.; Martin-Algarra, S. Phase II multicenter, single arm, open label study of nivolumab (NIVO) in combination with ipilimumab (IPI) as first line in adult patients (pts) with metastatic uveal melanoma (MUM): GEM1402 NCT02626962. J. Clin. Oncol. 2017, 35, 9533. [Google Scholar] [CrossRef]
- Scheulen, M.E.; Kaempgen, E.; Keilholz, U.; Heinzerling, L.; Ochsenreither, S.; Abendroth, A.; Hilger, R.A.; Grubert, M.; Wetter, A.; Guberina, N.; et al. STREAM: A randomized discontinuation, blinded, placebo-controlled phase II study of sorafenib (S) treatment of chemonaïve patients (pts) with metastatic uveal melanoma (MUM). J. Clin. Oncol. 2017, 35, 9511. [Google Scholar] [CrossRef]
- Patel, S.; Lewis, K.D.; Olencki, T.; Hernandez-Aya, L.; Joseph, R.; Williamson, S.; Chandra, S.; Shirai, K.; Moscow, J. A phase II study of glembatumumab vedotin for metastatic uveal melanoma. In Proceedings of the SMR Congress, Brisbane, Australia, 18–21 October 2017; p. 194. [Google Scholar]
- Sato, T.; Nathan, P.D.; Hernandez-Aya, L.; Sacco, J.J.; Orloff, M.M.; Visich, J.; Little, N.; Hulstine, A.-M.; Coughlin, C.M.; Carvajal, R.D. Redirected T cell lysis in patients with metastatic uveal melanoma with gp100-directed TCR IMCgp100: Overall survival findings. J. Clin. Oncol. 2018, 36, 9521. [Google Scholar] [CrossRef]
- Castet, F.; Garcia-Mulero, S.; Sanz-Pamplona, R.; Cuellar, A.; Casanovas, O.; Caminal, J.M.; Piulats, J.M. Uveal Melanoma, Angiogenesis and Immunotherapy, Is There Any Hope? Cancers 2019, 11, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.; Pilarski, R.; Boru, G.; Rehman, M.; Saqr, A.H.; Massengill, J.B.; Singh, A.; Marino, M.J.; Davidorf, F.H.; Cebulla, C.M.; et al. Germline BAP1 alterations in familial uveal melanoma. Genes. Chromosomes Cancer 2017, 56, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Pilarski, R.; Rai, K.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor Predisposition Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Decatur, C.L.; Ong, E.; Garg, N.; Anbunathan, H.; Bowcock, A.M.; Field, M.G.; Harbour, J.W. Driver Mutations in Uveal Melanoma: Associations With Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016, 134, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A. Rotterdam Ocular Melanoma Study Group Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.J.; Brouwer, N.J.; Esmaeli, B. The Cancer Genome Atlas Project: An Integrated Molecular View of Uveal Melanoma. Ophthalmology 2018, 125, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Trial of Niraparib in BAP1 and Other DNA Damage Response (DDR) Deficient Neoplasms (UF-STO-ETI-001). Available online: https://clinicaltrials.gov/ct2/show/NCT03207347 (accessed on 20 July 2020).
- Basile, M.S.; Mazzon, E.; Fagone, P.; Longo, A.; Russo, A.; Fallico, M.; Bonfiglio, V.; Nicoletti, F.; Avitabile, T.; Reibaldi, M. Immunobiology of Uveal Melanoma: State of the Art and Therapeutic Targets. Front. Oncol. 2019, 9, 1145. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Carlring, J.; Shaif-Muthana, M.; Sisley, K.; Rennie, I.G.; Murray, A.K. Apoptotic cell death in conjunction with CD80 costimulation confers uveal melanoma cells with the ability to induce immune responses. Immunology 2003, 109, 41–48. [Google Scholar] [CrossRef]
- Chen, P.W.; Mellon, J.K.; Mayhew, E.; Wang, S.; He, Y.G.; Hogan, N.; Niederkorn, J.Y. Uveal melanoma expression of indoleamine 2,3-deoxygenase: Establishment of an immune privileged environment by tryptophan depletion. Exp. Eye Res. 2007, 85, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Arguello, D.; Johnston, C.; Gatalica, Z.; Terai, M.; Weight, R.M.; Orloff, M.; Mastrangelo, M.J.; Sato, T. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 2017, 9, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Basile, M.S.; Mazzon, E.; Russo, A.; Mammana, S.; Longo, A.; Bonfiglio, V.; Fallico, M.; Caltabiano, R.; Fagone, P.; Nicoletti, F.; et al. Differential modulation and prognostic values of immune-escape genes in uveal melanoma. PLoS ONE 2019, 14, e0210276. [Google Scholar] [CrossRef]
- Harper, J.; Adams, K.J.; Bossi, G.; Wright, D.E.; Stacey, A.R.; Bedke, N.; Martinez-Hague, R.; Blat, D.; Humbert, L.; Buchanan, H.; et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoS ONE 2018, 13, e0205491. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Venzon, D.; White, D.; Rubin, J.T.; Lotze, M.T.; Simonis, T.B.; Balkissoon, J.; Rosenberg, S.A.; Parkinson, D.R. HLA association with response and toxicity in melanoma patients treated with interleukin 2-based immunotherapy. Cancer Res. 1992, 52, 6561–6566. [Google Scholar] [PubMed]
- ClinicalTrials.gov (NCT03070392). Safety and Efficacy of IMCgp100 Versus Investigator Choice in Advanced Uveal Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03070392 (accessed on 20 July 2020).
- Lopez, R.; Holyoke, E.D.; Moore, R.H.; Karakousis, C.P. Malignant melanoma with unknown primary site. J. Surg. Oncol. 1982, 19, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.; van der Veldt, A.; van Akkooi, A.; Verhoef, C.; Grünhagen, D.J.; Louwman, W.J. Treatment of melanoma of unknown primary in the era of immunotherapy and targeted therapy: A Dutch population-based study. Int. J. Cancer 2020, 146, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Choi, Y.Y.; Kim, D.S.; Lee, J.H.; Jang, H.S.; Lee, J.H.; Kim, H.; Oh, B.H.; Roh, M.R.; Nam, K.A.; et al. Metastatic melanomas of unknown primary show better prognosis than those of known primary: A systematic review and meta-analysis of observational studies. J. Am. Acad. Dermatol. 2015, 72, 59–70. [Google Scholar] [CrossRef]
- Gos, A.; Jurkowska, M.; van Akkooi, A.; Robert, C.; Kosela-Paterczyk, H.; Koljenović, S.; Kamsukom, N.; Michej, W.; Jeziorski, A.; Pluta, P.; et al. Molecular characterization and patient outcome of melanoma nodal metastases and an unknown primary site. Ann. Surg. Oncol. 2014, 21, 4317–4323. [Google Scholar] [CrossRef] [Green Version]
- Gambichler, T.; Chatzipantazi, M.; Schröter, U.; Stockfleth, E.; Gedik, C. Patients with melanoma of unknown primary show better outcome under immune checkpoint inhibitor therapy than patients with known primary: Preliminary results. Oncoimmunology 2019, 8, e1677139. [Google Scholar] [CrossRef] [Green Version]
- Perng, P.; Marcus, C.; Subramaniam, R.M. (18)F-FDG PET/CT and Melanoma: Staging, Immune Modulation and Mutation-Targeted Therapy Assessment, and Prognosis. Am. J. Roentgenol. 2015, 205, 259–270. [Google Scholar] [CrossRef]
- Henderson, F.; Johnston, H.R.; Badrock, A.P.; Jones, E.A.; Forster, D.; Nagaraju, R.T.; Evangelou, C.; Kamarashev, J.; Green, M.; Fairclough, M.; et al. Enhanced Fatty Acid Scavenging and Glycerophospholipid Metabolism Accompany Melanocyte Neoplasia Progression in Zebrafish. Cancer Res. 2019, 79, 2136–2151. [Google Scholar] [CrossRef] [Green Version]
- Faillace, W.J.; Okawara, S.H.; McDonald, J.V. Neurocutaneous melanosis with extensive intracerebral and spinal cord involvement. Report of two cases. J. Neurosurg. 1984, 61, 782–785. [Google Scholar] [CrossRef] [Green Version]
- Padilla-Vázquez, F.; Escobar-de la Garma, V.H.; Ayala-Arcipreste, A.; Mendizábal-Guerra, R.; Cuesta-Mejía, T. Melanocytoma and meningeal melanocytosis, similar but different lesions. Cirugía y Circ. 2017, 85, 273–278. [Google Scholar] [CrossRef]
- Garbacz, T.; Osuchowski, M.; Bartosik-Psujek, H. Primary diffuse meningeal melanomatosis—A rare form of meningeal melanoma: Case report. BMC Neurol. 2019, 19, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Pradhan, R.; Pal, S.; Bhattacharya, S.; Banerjee, A.; Bhattacharyya, D. Primary intracranial malignant melanoma in an adolescent girl: A case report. Clin. Cancer Investig. J. 2016, 5, 551–553. [Google Scholar]
- Zhang, S.; Wang, W.; Su, X.; Tan, Q.; Sun, H.; Liu, Z.; Chen, N.; Gong, Q.; Yue, Q. Amelanotic Meningeal Melanoma with Leptomeningeal Dissemination: A Case Report and Systematic Literature Review. World Neurosurg. 2019, 122, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Larson, T.C.; Houser, O.W.; Onofrio, B.M.; Piepgras, D.G. Primary spinal melanoma. J. Neurosurg. 1987, 66, 47–49. [Google Scholar] [CrossRef]
- Küsters-Vandevelde, H.V.N.; Germans, M.R.; Rabbie, R.; Rashid, M.; Ten Broek, R.; Blokx, W.A.M.; Prinsen, C.F.M.; Adams, D.J.; Ter Laan, M. Whole-exome sequencing of a meningeal melanocytic tumour reveals activating CYSLTR2 and EIF1AX hotspot mutations and similarities to uveal melanoma. Brain Tumor Pathol. 2018, 35, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Sakai, K.; Higashiyama, F.; Oya, F.; Maejima, T.; Miyake, T. Primary central nervous system malignant melanoma with leptomeningeal melanomatosis: A case report and review of the literature. Neurosurg. Rev. 2018, 41, 333–339. [Google Scholar] [CrossRef]
- El Habnouni, C.; Bléchet, C.; Bens, G. Pembrolizumab for primary malignant melanoma of the central nervous system. J. Neurooncol. 2018, 139, 225–227. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Cabral, E.S.; Kartha, R.V.; Swetter, S.M. Primary dermal melanoma: Distinct immunohistochemical findings and clinical outcome compared with nodular and metastatic melanoma. Arch. Dermatol. 2008, 144, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Teow, J.; Chin, O.; Hanikeri, M.; Wood, B.A. Primary dermal melanoma: A West Australian cohort. ANZ J. Surg. 2015, 85, 664–667. [Google Scholar] [CrossRef]
- Harris, C.G.; Lo, S.; Ahmed, T.; Scolyer, R.A.; Ferguson, P.M.; Karim, R.Z.; Lam, T.K.; Lee, K.K.; Shannon, K.F.; Spillane, A.J.; et al. Primary dermal melanoma: Clinical behaviour, prognosis and treatment. Eur. J. Surg. Oncol. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, K.; Kato, Y.; Kanno, J.; Kasuga, T. Melanocytes and melanosis of the oesophagus in Japanese subjects—Analysis of factors effecting their increase. Virchows Arch. A Pathol. Anat. Histopathol. 1990, 417, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Schizas, D.; Mylonas, K.S.; Bagias, G.; Mastoraki, A.; Ioannidi, M.; Kanavidis, P.; Hasemaki, N.; Karavokyros, I.; Theodorou, D.; Liakakos, T. Esophageal melanoma: A systematic review and exploratory recurrence and survival analysis. Dis. Esophagus 2019, 32, doz083. [Google Scholar] [CrossRef]
- Caldwell, C.B.; Bains, M.S.; Burt, M. Unusual malignant neoplasms of the esophagus. Oat cell carcinoma, melanoma, and sarcoma. J. Thorac. Cardiovasc. Surg. 1991, 101, 100–107. [Google Scholar] [CrossRef]
- Gao, S.; Li, J.; Feng, X.; Shi, S.; He, J. Characteristics and Surgical Outcomes for Primary Malignant Melanoma of the Esophagus. Sci. Rep. 2016, 6, 23804. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Makino, T.; Yamasaki, M.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Motoori, M.; Kimura, Y.; Nakajima, K.; et al. Clinicopathological characteristics and survival of primary malignant melanoma of the esophagus. Oncol. Lett. 2019, 18, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- HE, Y.; MOU, J.; LUO, D.; GAO, B.; WEN, Y. Primary malignant melanoma of the breast: A case report and review of the literature. Oncol. Lett. 2014, 8, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; Ehlers, R.A.; Hunt, K.K.; Yi, M.; Kuerer, H.M.; Singletary, S.E.; Ross, M.I.; Feig, B.W.; Symmans, W.F.; Meric-Bernstam, F. Metastases to the breast from nonbreast solid neoplasms: Presentation and determinants of survival. Cancer 2007, 110, 731–737. [Google Scholar] [CrossRef]
- Rassouli, M.; Voutsadakis, I.A. Primary Noncutaneous Malignant Melanoma of the Breast. Breast J. 2016, 22, 688–691. [Google Scholar] [CrossRef]
- Koh, J.; Lee, J.; Jung, S.Y.; Kang, H.S.; Yun, T.; Kwon, Y. Primary Malignant Melanoma of the Breast: A Report of Two Cases. J. Pathol. Transl. Med. 2019, 53, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Sun, X.; Zhu, Z.; Lu, J.; Wen, G.; Chang, X.; Gao, H.; Hua, Y.; Wang, L.; Gao, J. Primary malignant melanoma of the lung: A case report and literature review. BMC Pulm. Med. 2020, 20, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gómez Tejeda Zañudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef]
- Kee, D.; Kondrashova, O.; Ananda, S.; Brown, M.P.; Cohen, P.A.; Dean, A.; Desai, J.; Fellowes, A.; Fox, S.B.; Hadley, A.; et al. NOMINATOR: Feasibility of genomic testing of rare cancers to match cancer to treatment. J. Clin. Oncol. 2020, 38, 103. [Google Scholar] [CrossRef]
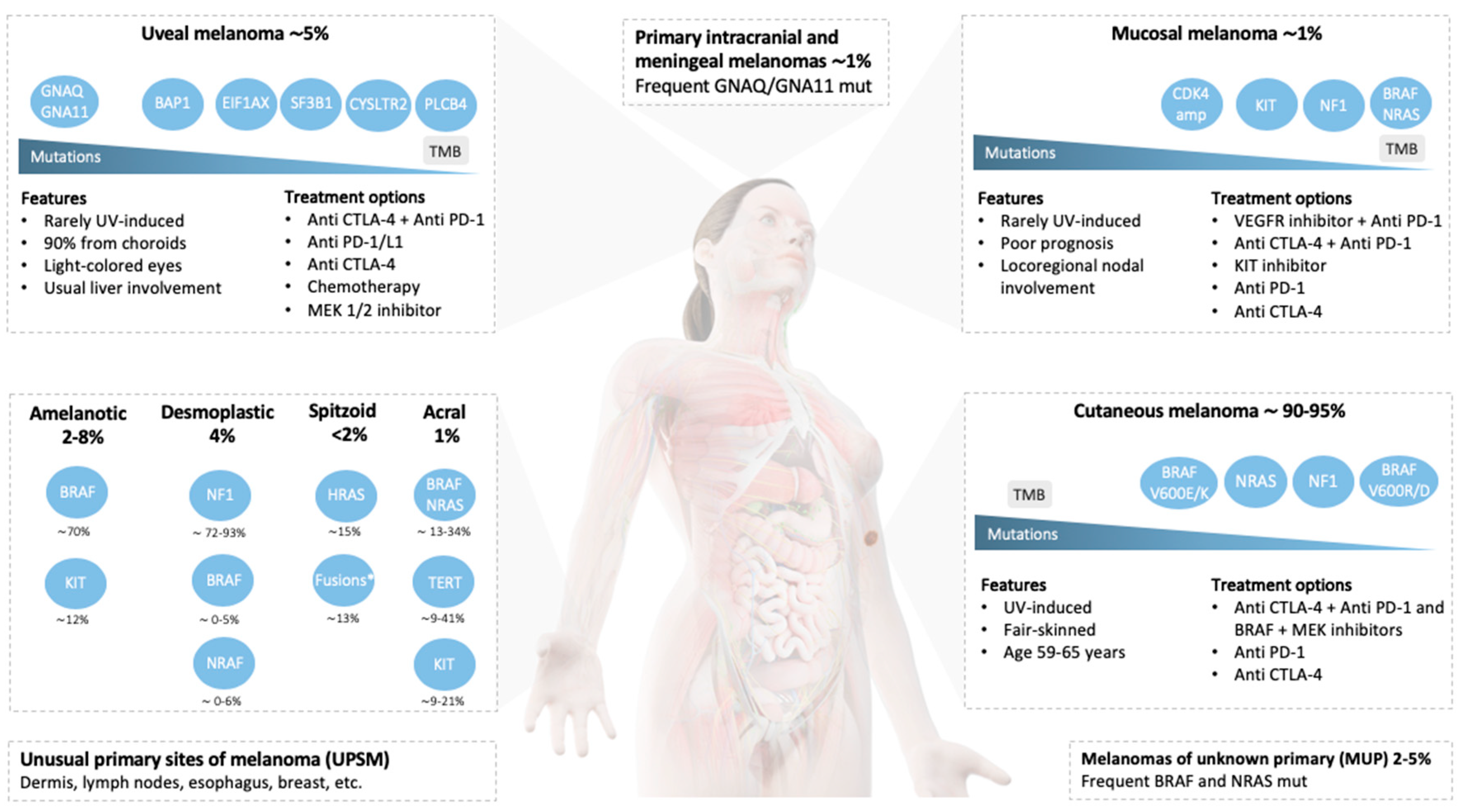

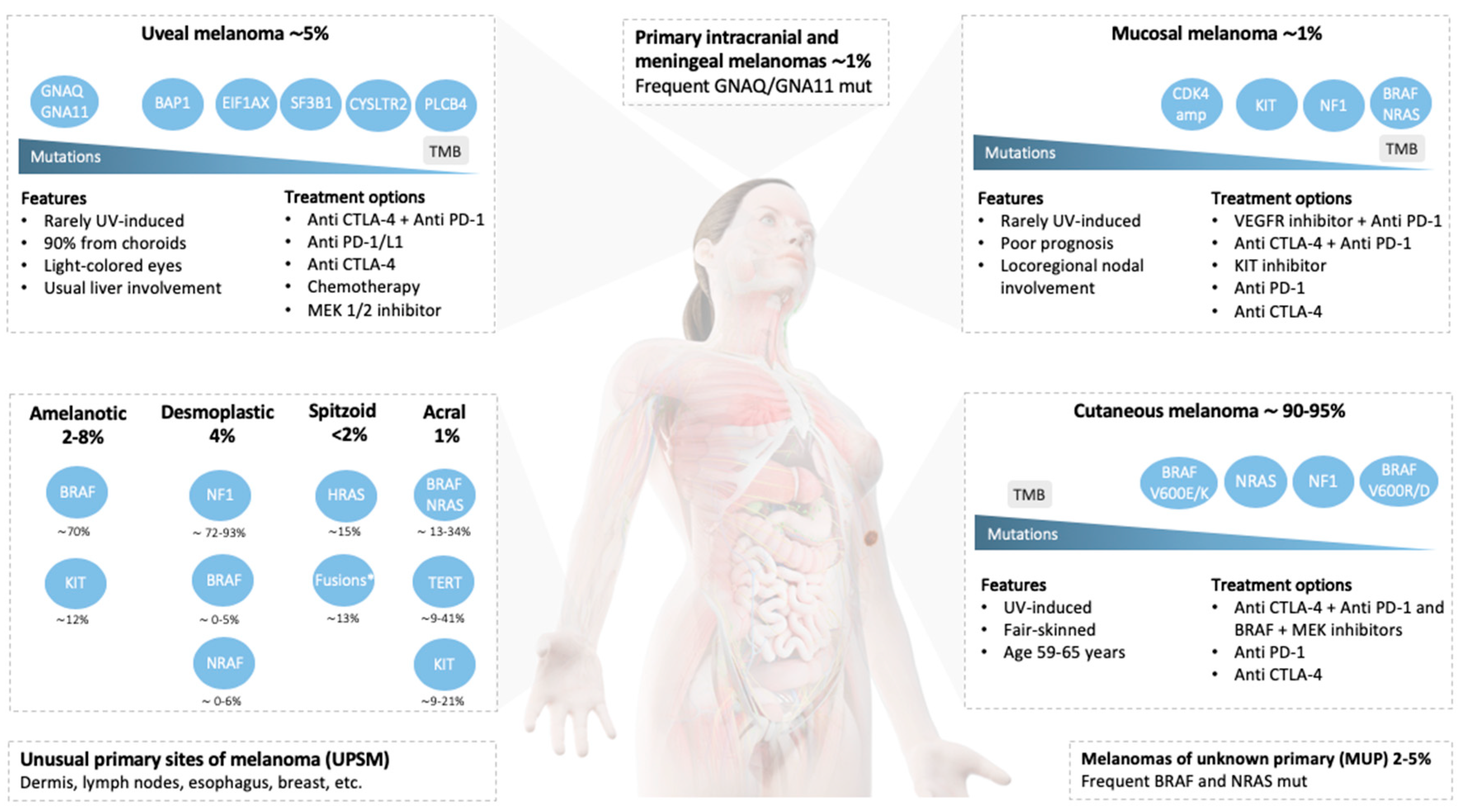{kind=link}
| Study | Phase | N | Arms | ORR (%) | DCR (%) | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|
| Dummer et al. 2017 [18] | 3 a,e | 402 | Binimetinib; Dacarbazine | 15; 7 | 58; 25 | 2.8; 1.5 | 11; 10.1 |
| Lebbe et al. 2016 [19] | 2 b,e | 194 | Pimasertib; Dacarbazine | 27; 14 | 33; 16 | 3.3; 1.7 | 8.9; 10.6 |
| Ascierto et al. 2013 [20] | 2 d | 30 | Binimetinib | 10 | 63 | 3.7 | NS |
| Kirkwood et al. 2012 [21] | 2 c,e | 10; 18 | Selumetinib; Temozolomide | 0; 6 | 50; 55 | NS | NS |
| Kim et al. 2019 [22] | 1 e | 9 | Belvarafenib | 44 | NS | 6.2 | NS |
| Schuller et al. 2017 [23] | 1b e | 16 | Ribociclib + Binimetinib | 25 | 69 | 6.7 | NS |
| Algazi et al. 2017 [24] | 1 d,e | 10 | GSK2141795 f + Trametinib | 0 | 40 | 2.3 | 4 |
| Sullivan et al. 2017 [25] | 1 e | 18 | Ulixertinib | 17 | NS | NS | NS |
| Falchook et al. 2012 [26] | 1 b,e | 7 | Trametinib | 0 | 22 | NS | NS |
| Study | Phase | N | Subtype (n) | Arms | ORR (%) | DCR (%) | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|---|
| Kim et al. 2008 [86] | 2 b | 21 | Cutaneous (7) Acral (2) Soft part (1) Unclassified (11) | Imatinib | 4.8 | 23.8 | 1.4 | 7.5 |
| Guo et al. 2011 [87] | 2 a | 43 | Acral (21) Mucosal (11) Cutanous (9) Unknown (2) | Imatinib | 23.3 | 53.5 | 3.5 | 14 |
| Carvajal et al. 2011 [88] | 2a | 28 | Mucosal (13) Acral (10) Cutaneous (5) | Imatinib | Mucosal 23 Acral 38 Cutaneous 0 | NS | 2.8 | 10.7 |
| Hodi et al. 2013 [89] | 2 a | 24 | Mucosal (17) Acral (6) Cutenous (1) | Imatinib | 29 | 50 | 3.7 | 12.5 |
| Cho et al. 2012 [90] | 2 a | 11 | Acral (9) Mucosal (2) | Nilotinib | 22.2 | 77.8 | 2.5 | 7.7 |
| Carvajal et al. 2015 [91] | 2 a,e | 19 | Mucosal (12) Acral (4) Cutaneous (3) | Nilotinib | Mucosal 27.2 Acral 0 Cutaneous 0 | Mucosal 63.6 Acral 25 Cutaneous 33.3 | 3.4 f 2.6 g | 14.2 f 4.3 g |
| Lee et al. 2015 [92] | 2 b | 27 | Acral (15) Mucosal (7) Cutaneous (5) | Nilotinib | Acral 40 Mucosal 0 Cutaneous 0 | Acral 73.3 Mucosal 28.6 Cutaneous 40 | NS | NS |
| Guo et al. 2017 [93] | 2 b | 42 | Acral (20) Mucosal (20) Cutaneous (2) | Nilotinib | Acral 25 Mucosal 25 Cutaneous 50 | Acral 80 Mucosal 70 Cutaneous 50 | 4.2 | 18 |
| Deylon et al. 2018 [94] | 2 b | 22 | Mucosal (9) Acral (7) Cutaneous (6) | Nilotinib | Mucosal 33.3 Acral 14.3 Cutaneous 16.6 | Mucosal 66.6 Acral 71.4 Cutaneous 80 | 6 d | 13.2 d |
| Kalinsky et al. 2016 [95] | 2 b | 25 | Acral (15) Mucosa (10) | Dasatinib | Acral 33 Mucosal 14 | 50 c | 2.7 | 11.8 |
| Minor et al. 2012 [96] | - b | 6 | Mucosal (NS) Acral (NS) Cutaneous (NS) | Sunitinib | Mucosal 60 Acral 0 | Mucosal 60 Acral 0 | NS | NS |
| Study | Study Type | N | Arms (n) | ORR (%) | DCR (%) | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|
| Postow et al. 2013 [150] | Multicenter, retrospective | 33 | Ipilimumab | 6.7 | 26.7 | NS | 6.4 |
| Del Vecchio et al. 2014 [151] | Expanded, access program | 71 | Ipilimumab | 11 | 36.2 | 4.3 | 6.4 |
| Shoushtari et al. 2016 [100] | Multi-institutional, retrospective | 35 | Nivolumab or Pembrolizumab | 23 | 42.9 | 3.9 | NS |
| D’Angelo et al. 2017 [141] | Pooled analysis of phase 1-2-3 studies b | 157 | Nivolumab + Ipilimumab (86) Nivolumab (35) Ipilimumab (36) | 37.1 23.3 8.3 | 57.1 45.3 16.7 | 5.9 3 2.7 | NS |
| Mignard et al. 2018 [138] | Multicenter, retrospective | 151 | Ipilimumab (76) Nivolumab or Pembrolizumab (75) | 11.9 | 17.9 | 15.97 | NS |
| Omid et al. 2019 [139] | Post-hoc analysis of phase 1-2-3 studies a | 84 | Pembrolizumab | 19 | 31 | 2.8 | 11.3 |
| Moya-Plana et al. 2019 [140] | Single-center prospective cohort | 44 | Ipilimumab (24) Pembrolizumab (20) | 8.2 35 | 30 45 | 3 5 | 12 16.2 |
| Si Lu et al. 2019 [152] | Phase 1b | 15 | Pembrolizumab | 13.3 | 20 | NS | NS |
| Shoushtari et al. 2020 [142] | Subgroup of CheckMate 067 | 79 | Ipilimumab + Nivolumab (28) Nivolumab (23) Ipilimumab (28) | 43 30 7 | 57 39 11 | 5.8 3.0 2.6 | 22.7 20.2 12.1 |
| Sheng et al. 2020 [148] | Phase 1b | 29 | Axitinib + Toripalimab | 48.5 | 84.8 | 7.5 | 20.7 |
| Si Lu et al. 2020 [149] | Phase 2 | 40 | Vorolanib + Toripalimab | 15–22.2 | 55.5–65 | 5.6–5.7 | NS |
| Study | Study Type | N | Arms (n) | ORR (%) | DCR (%) | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|
| Luke et al. 2013 [172] | Multicenter, retrospective | 39 | Ipilimumab | 2.6 | 46 | - | 9.6 |
| Piulats et al. 2014 [173] | Phase 2 | 32 | Ipilimumab | 6.45 | 50 | NS | NS |
| Carvajal et al. 2014 [169] | Phase 2 | 101 | Selumetinib (50) Chemotherapy (51) | 14 0 | NS | 3.7 1.6 | 11.8 9.1 |
| Zimmer et al. 2015 [174] | Phase 2 | 53 | Ipilimumab | 0 | 47 | 2.8 | 6.8 |
| Joshua et al. 2015 [175] | Phase 2 | 11 | Tremelimumab | 0 | - | 2.9 | 12.8 |
| Shoushtari et al. 2016 [171] | Phase 2 | 39 | Trametinib (18) Trametinib + GSK2141795 a (21) | 5.5 4.8 | NS | 3.6 3.6 | NS |
| Tsai et al. 2016 [176] | Multicenter, retrospective | 58 | Pembrolizumab (40) Nivolumab (16) Atezolizumab (2) | 3 | 10 | 2.7 | 9.5 |
| Piulats et al. 2017 [177] | Phase 2 | 19 | Nivolumab + Ipilimumab | 15.8 | 63.2 | 4.99 | NR |
| Scheulen et al. 2017 [178] | Phase 2 | 118 | Sorafenib | 1.7 | 66.1 | 5.5 | 14.8 |
| Patel et al. 2017 [179] | Phase 2 | 31 | Glembatumumab Vedotin b | 6 | 61 | 3.2 | 11.8 |
| Mignard et al. 2018 [138] | Multicenter, Retrospective | 100 | Ipilimumab (63) Nivolumab or Pembrolizumab (37) | 0 | 32 | - | 13.38 |
| Carvajal et al. 2018 [170] | Phase 3 | 129 | Selumetinib + Dacarbazine (97) Placebo + Dacarbazine (32) | 3 0 | NS | 2.8 1.8 | NS |
| Sato et al. 2018 [180] | Phase 1/2 | 19 | Tebentafusp c | 10.5 d | - | - | NR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacón, M.; Pfluger, Y.; Angel, M.; Waisberg, F.; Enrico, D. Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives. Cancers 2020, 12, 2362. https://doi.org/10.3390/cancers12092362
Chacón M, Pfluger Y, Angel M, Waisberg F, Enrico D. Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives. Cancers. 2020; 12(9):2362. https://doi.org/10.3390/cancers12092362
Chicago/Turabian StyleChacón, Matías, Yanina Pfluger, Martín Angel, Federico Waisberg, and Diego Enrico. 2020. "Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives" Cancers 12, no. 9: 2362. https://doi.org/10.3390/cancers12092362
APA StyleChacón, M., Pfluger, Y., Angel, M., Waisberg, F., & Enrico, D. (2020). Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives. Cancers, 12(9), 2362. https://doi.org/10.3390/cancers12092362