Dual Inhibition of Autophagy and PI3K/AKT/MTOR Pathway as a Therapeutic Strategy in Head and Neck Squamous Cell Carcinoma


 , , , and
, , , and
Abstract
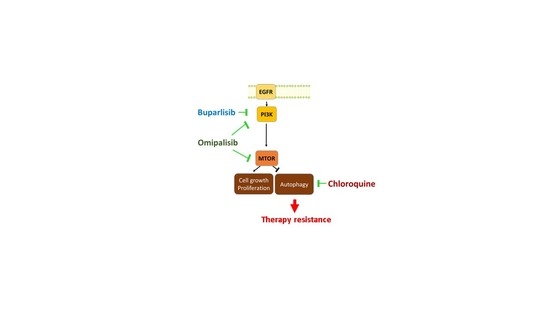
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PI3Ki Induce Autophagy Flux in HNSCC Cell Lines
2.2. PI3Ki and CQ Work in Synergy to Decrease Proliferation of HNSCC Cell Lines
2.3. CQ Inhibits PI3Ki-Induced Autophagy
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. IncuCyte® Live-Cell Analysis for Cell Proliferation and Bliss Synergy Score
4.3. Bliss Synergy Score Calculation
4.4. Acridine Orange Staining
4.5. Immunoblotting
4.6. Tandem Sensor RFP-GFP-LC3B Expression
4.7. Reagents
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 2010, 15, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef]
- Wirtz, E.D.; Hoshino, D.; Maldonado, A.T.; Tyson, D.R.; Weaver, A.M. Response of head and neck squamous cell carcinoma cells carrying PIK3CA mutations to selected targeted therapies. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Dodhia, S.; Su, G.H. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef] [Green Version]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.; van der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [Green Version]
- Soulieres, D.; Licitra, L.; Mesia, R.; Remenar, E.; Li, S.H.; Karpenko, A.; Chol, M.; Wang, Y.A.; Solovieff, N.; Bourdeau, L.; et al. Molecular Alterations and Buparlisib Efficacy in Patients with Squamous Cell Carcinoma of the Head and Neck: Biomarker Analysis from BERIL-1. Clin. Cancer Res. 2018, 24, 2505–2516. [Google Scholar] [CrossRef] [Green Version]
- Soulieres, D.; Faivre, S.; Mesia, R.; Remenar, E.; Li, S.H.; Karpenko, A.; Dechaphunkul, A.; Ochsenreither, S.; Kiss, L.A.; Lin, J.C.; et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017, 18, 323–335. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, M.; Dieude, M.; Yang, B.; Hamelin, K.; Underwood, K.; Hebert, M.J. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 2014, 10, 2193–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.F.; Bu, L.L.; Wang, W.M.; Ma, S.R.; Liu, J.F.; Deng, W.W.; Mao, L.; Yu, G.T.; Huang, C.F.; Liu, B.; et al. Tumor growth suppression by inhibiting both autophagy and STAT3 signaling in HNSCC. Oncotarget 2015, 6, 43581–43593. [Google Scholar] [CrossRef] [PubMed]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhao, X.; Lang, L.; Shay, C.; Andrew Yeudall, W.; Teng, Y. Autophagy blockade sensitizes human head and neck squamous cell carcinoma towards CYT997 through enhancing excessively high reactive oxygen species-induced apoptosis. J. Mol. Med. 2018, 96, 929–938. [Google Scholar] [CrossRef]
- Xie, J.; Li, Q.; Ding, X.; Gao, Y. Targeting mTOR by CZ415 Inhibits Head and Neck Squamous Cell Carcinoma Cells. Cell Physiol. Biochem. 2018, 46, 676–686. [Google Scholar] [CrossRef]
- Chang, I.; Wang, C.Y. Inhibition of HDAC6 Protein Enhances Bortezomib-induced Apoptosis in Head and Neck Squamous Cell Carcinoma (HNSCC) by Reducing Autophagy. J. Biol. Chem. 2016, 291, 18199–18209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Peng, Y.; Tang, H.; He, X.; Wang, Z.; Hu, D.; Zhou, X. Autophagy induction contributes to GDC-0349 resistance in head and neck squamous cell carcinoma (HNSCC) cells. Biochem. Biophys. Res. Commun. 2016, 477, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Sun, M.; Ge, X.; Sun, Y. EGFR tyrosine kinase inhibitors differentially affect autophagy in head and neck squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2017, 486, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kansy, B.A.; Li, J.; Cong, L.; Liu, Y.; Trivedi, S.; Wen, H.; Ting, J.P.; Ouyang, H.; Ferris, R.L. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene 2016, 35, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Sannigrahi, M.K.; Singh, V.; Sharma, R.; Panda, N.K.; Khullar, M. Role of autophagy in head and neck cancer and therapeutic resistance. Oral Dis. 2015, 21, 283–291. [Google Scholar] [CrossRef]
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, e3. [Google Scholar] [CrossRef]
- Levy, J.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermed. Sci. 2017, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, F.; Thomas, C.; Crafter, C.; Kumano, M.; Zhang, F.; Davies, B.R.; Gleave, M.E.; Zoubeidi, A. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin. Cancer Res. 2013, 19, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortnik, S.; Gorski, S.M. Clinical Applications of Autophagy Proteins in Cancer: From Potential Targets to Biomarkers. Int. J. Mol. Sci. 2017, 18, 1496. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Ma, C.X.; Luo, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; Griffith, M.; Griffith, O.L.; Skidmore, Z.L.; Spies, N.C.; Ramu, A.; et al. A Phase I Trial of BKM120 (Buparlisib) in Combination with Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 1801992. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Supko, J.G.; Neal, J.W.; Muzikansky, A.; Digumarthy, S.; Fidias, P.; Temel, J.S.; Heist, R.S.; Shaw, A.T.; McCarthy, P.O.; et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Pascolo, S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur. J. Pharmacol. 2016, 771, 139–144. [Google Scholar] [CrossRef]
- Mazumdar, T.; Byers, L.A.; Ng, P.K.; Mills, G.B.; Peng, S.; Diao, L.; Fan, Y.H.; Stemke-Hale, K.; Heymach, J.V.; Myers, J.N.; et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2738–2750. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisa, L.; Barras, D.; Medova, M.; Aebersold, D.M.; Medo, M.; Poliakova, M.; Koch, J.; Bojaxhiu, B.; Elicin, O.; Dettmer, M.S.; et al. Comprehensive Genomic Profiling of Patient-matched Head and Neck Cancer Cells: A Preclinical Pipeline for Metastatic and Recurrent Disease. Mol. Cancer Res. 2018, 16, 1912–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginkel, J.H.; de Leng, W.W.; de Bree, R.; van Es, R.J.; Willems, S.M. Targeted sequencing reveals TP53 as a potential diagnostic biomarker in the post-treatment surveillance of head and neck cancer. Oncotarget 2016, 7, 61575–61586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Bansal, M.; Yang, J.; Karan, C.; Menden, M.P.; Costello, J.C.; Tang, H.; Xiao, G.; Li, Y.; Allen, J.; Zhong, R.; et al. A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol. 2014, 32, 1213–1222. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Caponio, V.C.A.; Troiano, G.; Adipietro, I.; Zhurakivska, K.; Arena, C.; Mangieri, D.; Mascitti, M.; Cirillo, N.; Lo Muzio, L. Computational analysis of TP53 mutational landscape unveils key prognostic signatures and distinct pathobiological pathways in head and neck squamous cell cancer. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Simeone, K.; Guay-Lord, R.; Lateef, M.A.; Peant, B.; Kendall-Dupont, J.; Orimoto, A.M.; Carmona, E.; Provencher, D.; Saad, F.; Gervais, T.; et al. Paraffin-embedding lithography and micro-dissected tissue micro-arrays: Tools for biological and pharmacological analysis of ex vivo solid tumors. Lab Chip 2019, 19, 693–705. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, M.; Cardin, G.B.; Cahuzac, M.; Ayad, T.; Bissada, E.; Guertin, L.; Bahig, H.; Nguyen-Tan, P.F.; Filion, E.; Ballivy, O.; et al. Dual Inhibition of Autophagy and PI3K/AKT/MTOR Pathway as a Therapeutic Strategy in Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2371. https://doi.org/10.3390/cancers12092371
Bernard M, Cardin GB, Cahuzac M, Ayad T, Bissada E, Guertin L, Bahig H, Nguyen-Tan PF, Filion E, Ballivy O, et al. Dual Inhibition of Autophagy and PI3K/AKT/MTOR Pathway as a Therapeutic Strategy in Head and Neck Squamous Cell Carcinoma. Cancers. 2020; 12(9):2371. https://doi.org/10.3390/cancers12092371
Chicago/Turabian StyleBernard, Monique, Guillaume B. Cardin, Maxime Cahuzac, Tareck Ayad, Eric Bissada, Louis Guertin, Houda Bahig, Phuc Felix Nguyen-Tan, Edith Filion, Olivier Ballivy, and et al. 2020. "Dual Inhibition of Autophagy and PI3K/AKT/MTOR Pathway as a Therapeutic Strategy in Head and Neck Squamous Cell Carcinoma" Cancers 12, no. 9: 2371. https://doi.org/10.3390/cancers12092371