An Integrative Omics Approach Reveals Involvement of BRCA1 in Hepatic Metastatic Progression of Colorectal Cancer

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Colorectal Cancer Cells
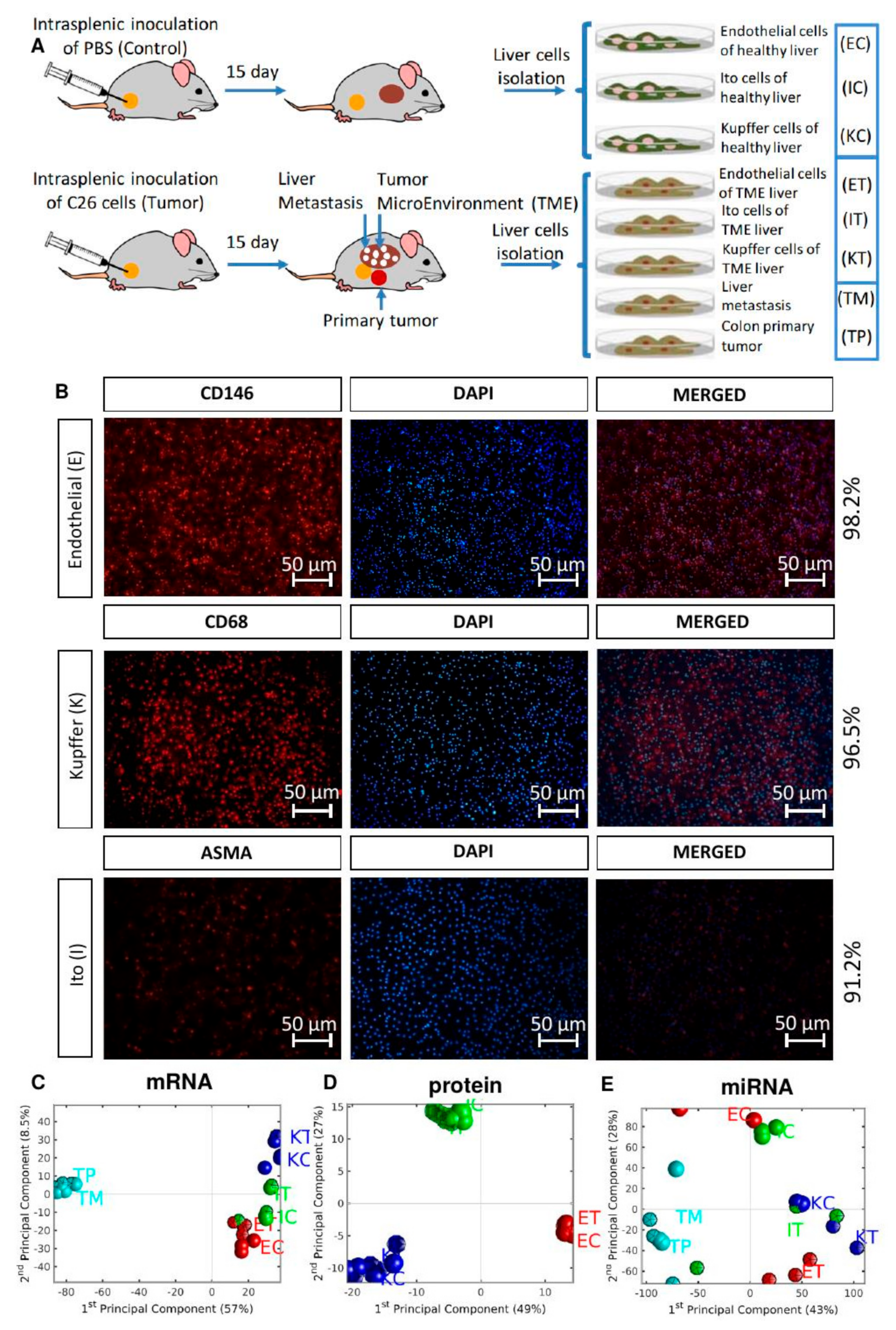
2.3. Control and Tumor-Activated Hepatic Cell Isolation and Culture
2.4. Omics Analysis
2.5. miRNA Validation with miRNA RT-qPCR
2.6. Protein-Gene Correlation Analysis
2.7. Algorithm to Search for miRNAs Targeting Genes
2.8. Gene-miRNA-Protein Network Centered in Brca1
2.9. CRC Patients and Samples
2.10. BRCA1 Immunostaining of TMA
3. Results
3.1. The Global Differences between Control and TME Samples are Most Pronounced at miRNA Expression Level
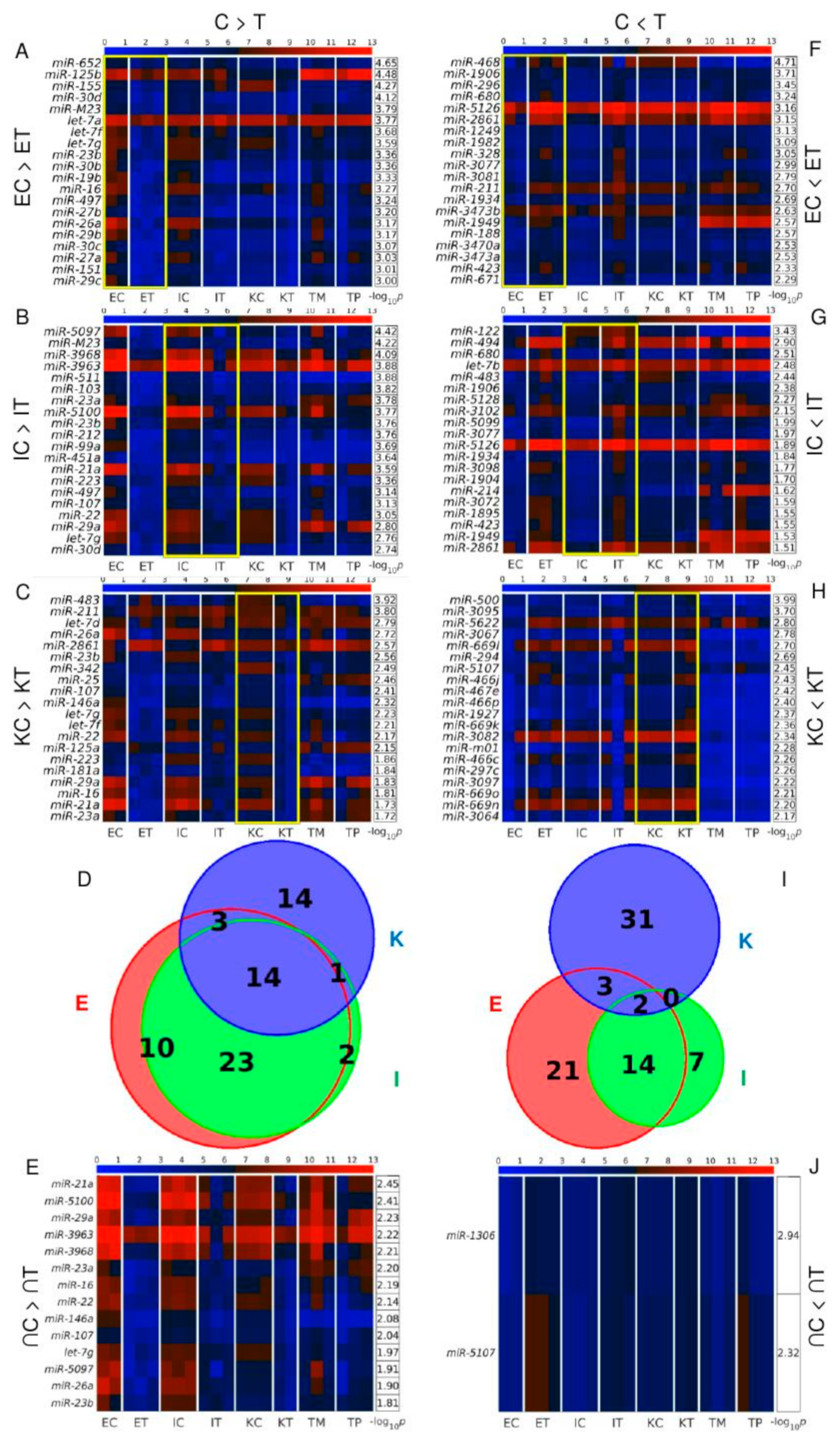
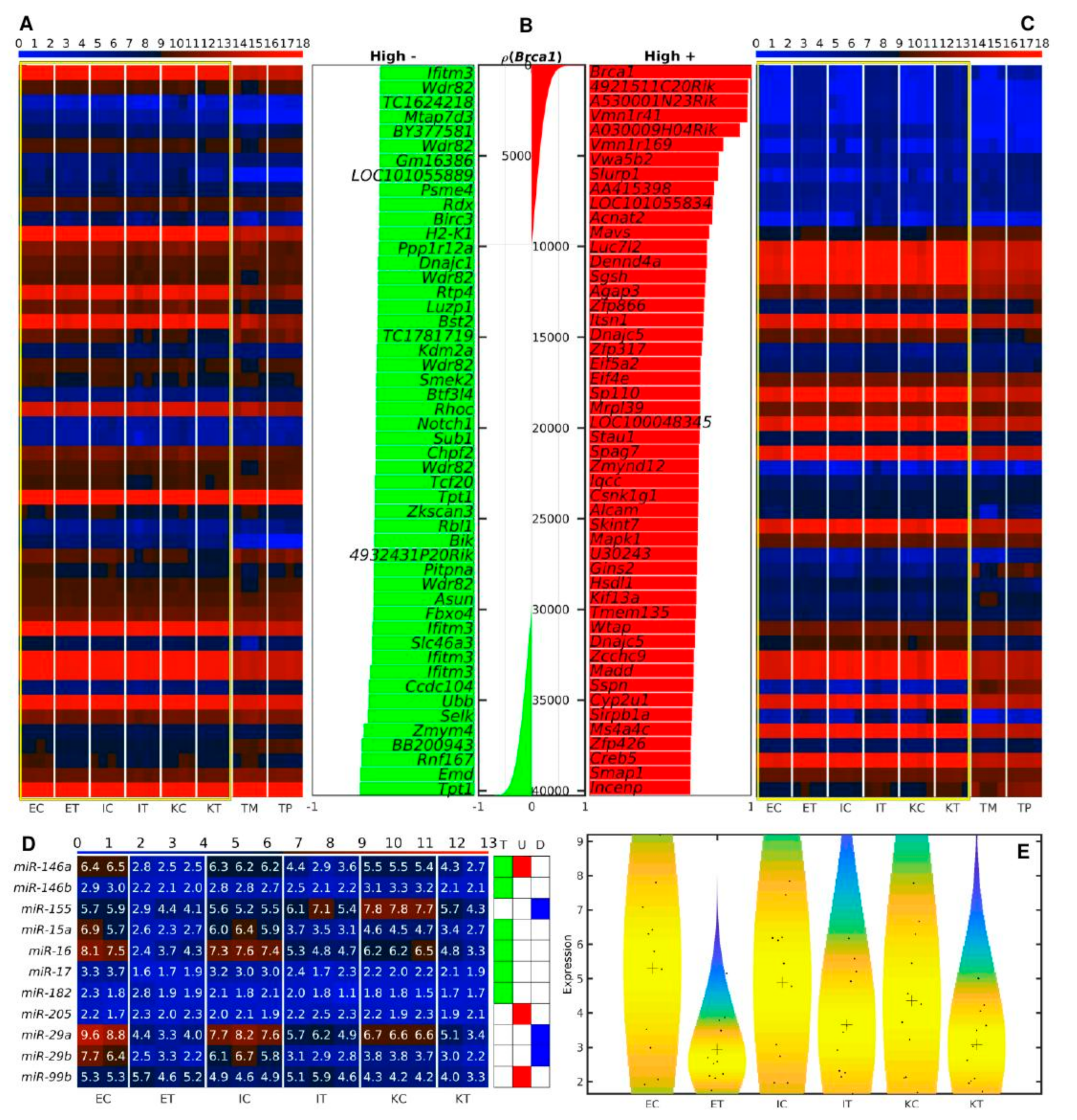
3.2. miR-21a, miR-146a, miR-16 and miR-29a Are among the miRNAs Simultaneously Down-Regulated in All the TME Cells
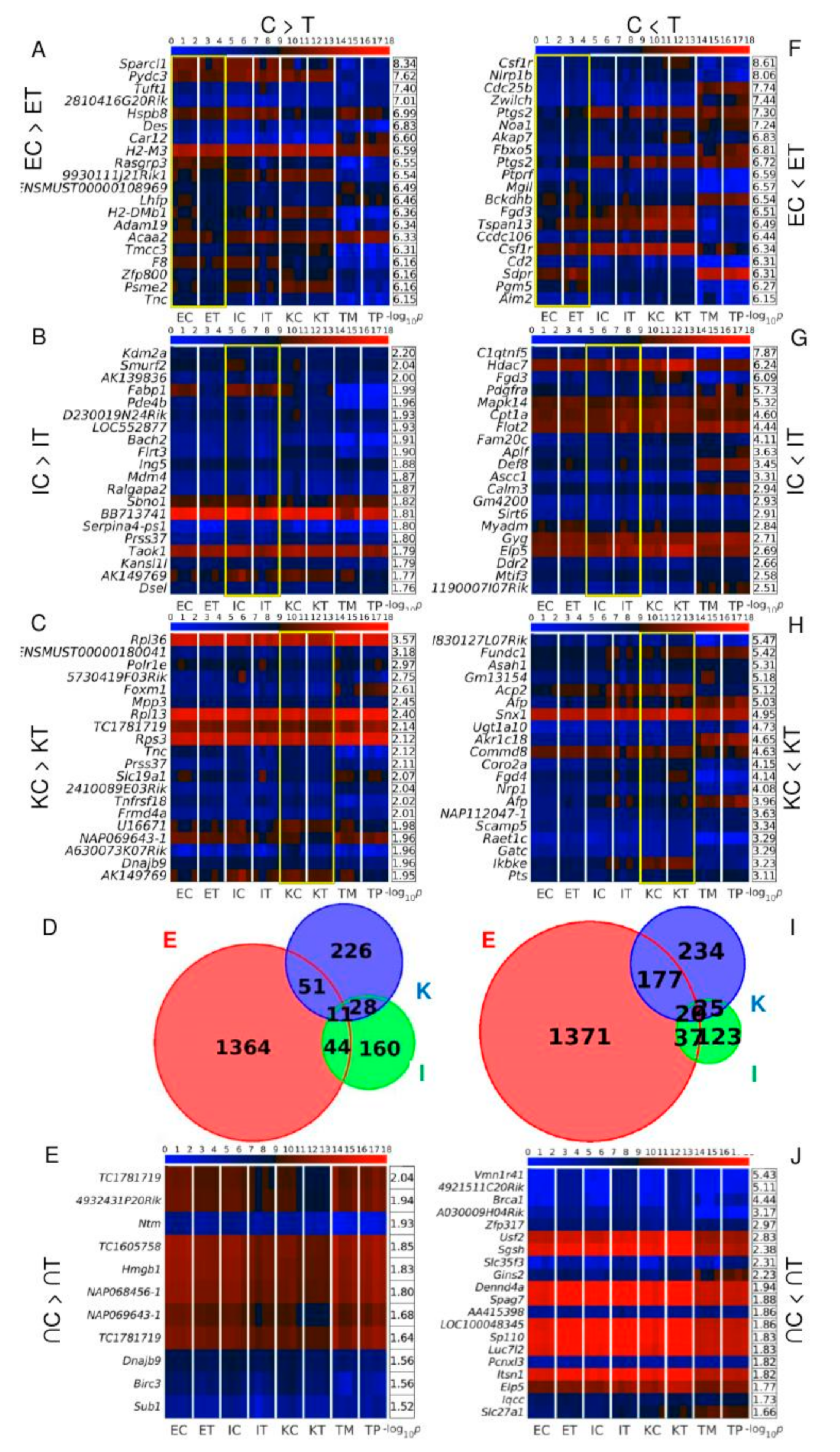
3.3. Brca1 and Sp110 Genes Are among the Twenty Transcripts Simultaneously Up-Regulated in All the TME Cells
3.4. Slurp1 and Acnat2 Are Highly Positively, and Tpt1, Emd and Rnf167 Are the Top Genes Highly Negatively Correlated with Brca1 across All Samples
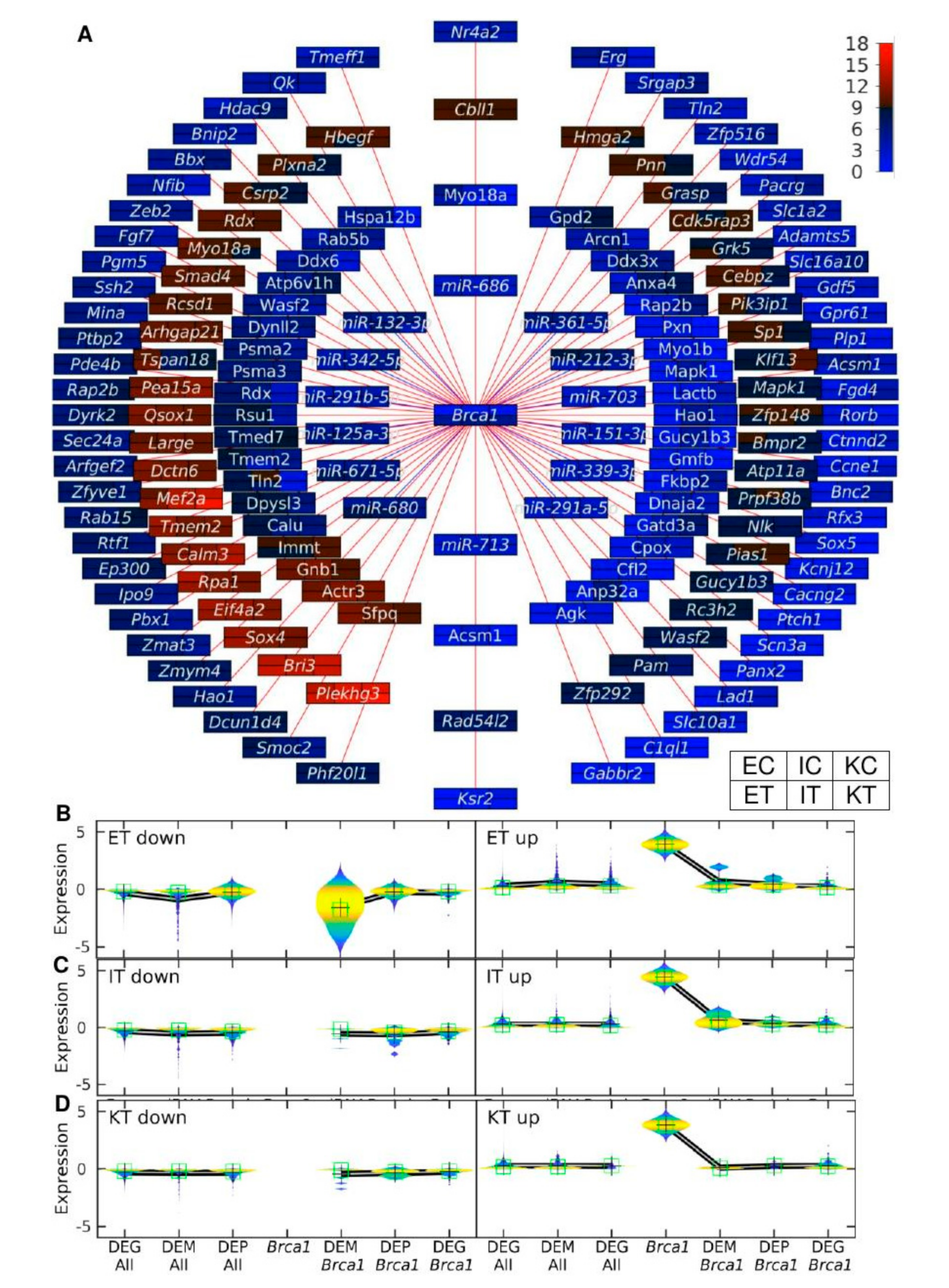
3.5. The Brca1-Centered Gene-miRNA-Protein Network Shows miR-212-3p as Simultaneously Down-Regulated in Tumor-Colonized Samples
3.6. RT-PCR Analysis Validated that the Brca1-Related miR-212-3p Is Differentially Expressed between Control and Tumor-Colonized Samples
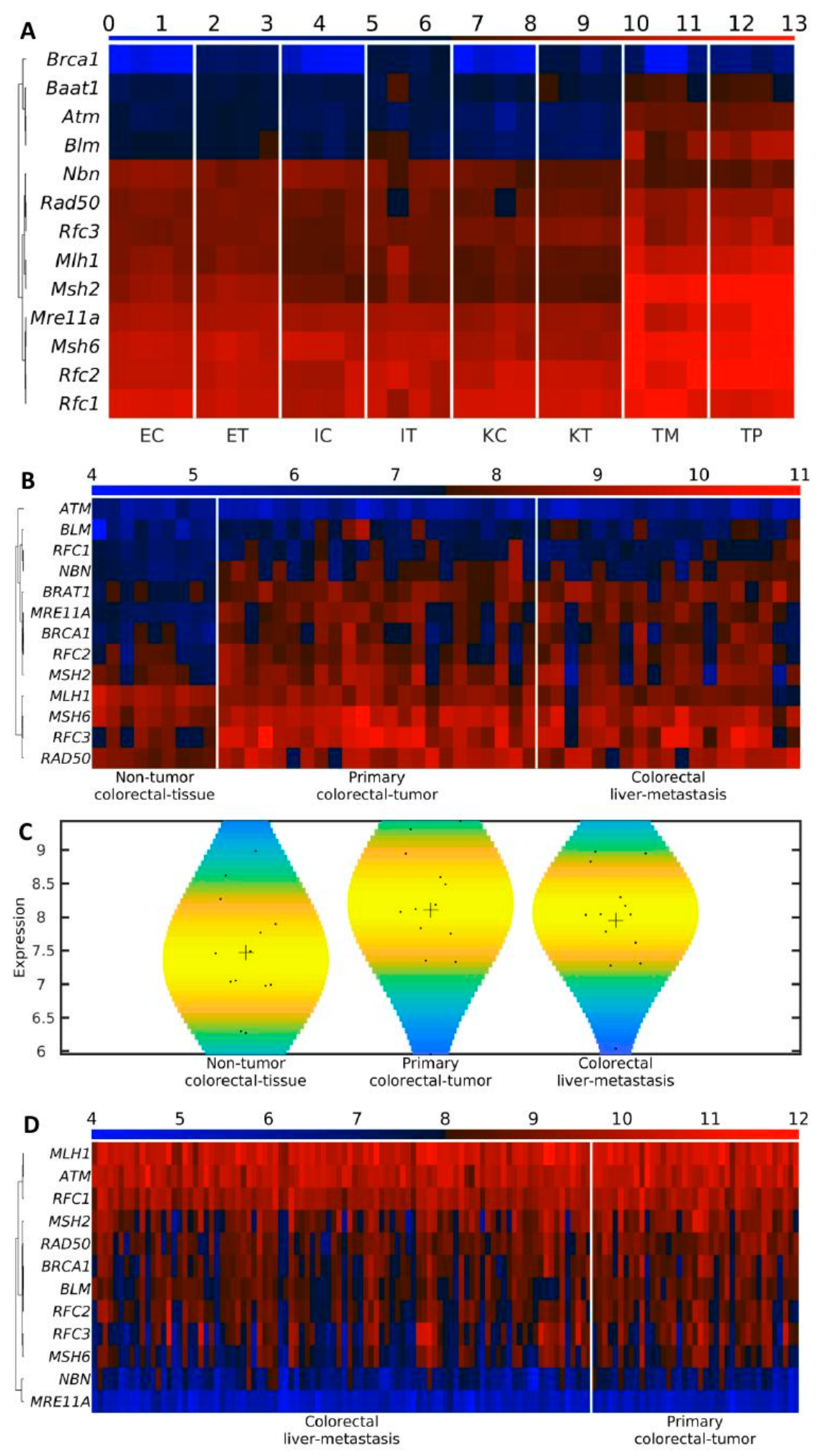
3.7. BRCA1 Is the Last BASC Gene Activated in the TME
3.8. The Most Probable Sequence of Cell Activation during Metastasis is Endothelial→Ito→Kupffer
3.9. Multiple Inflammation-Related Genes Are Up-Regulated in TME Samples
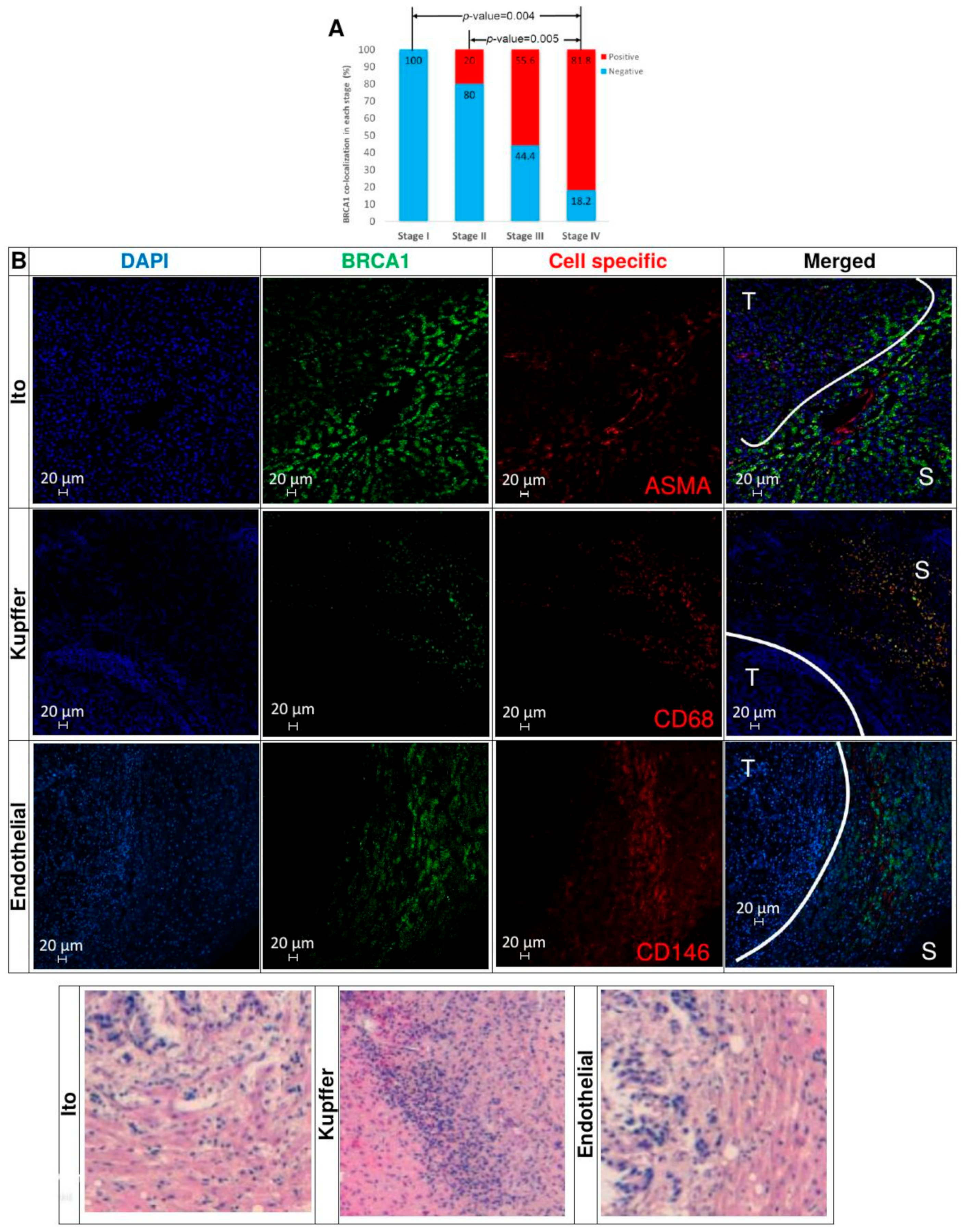
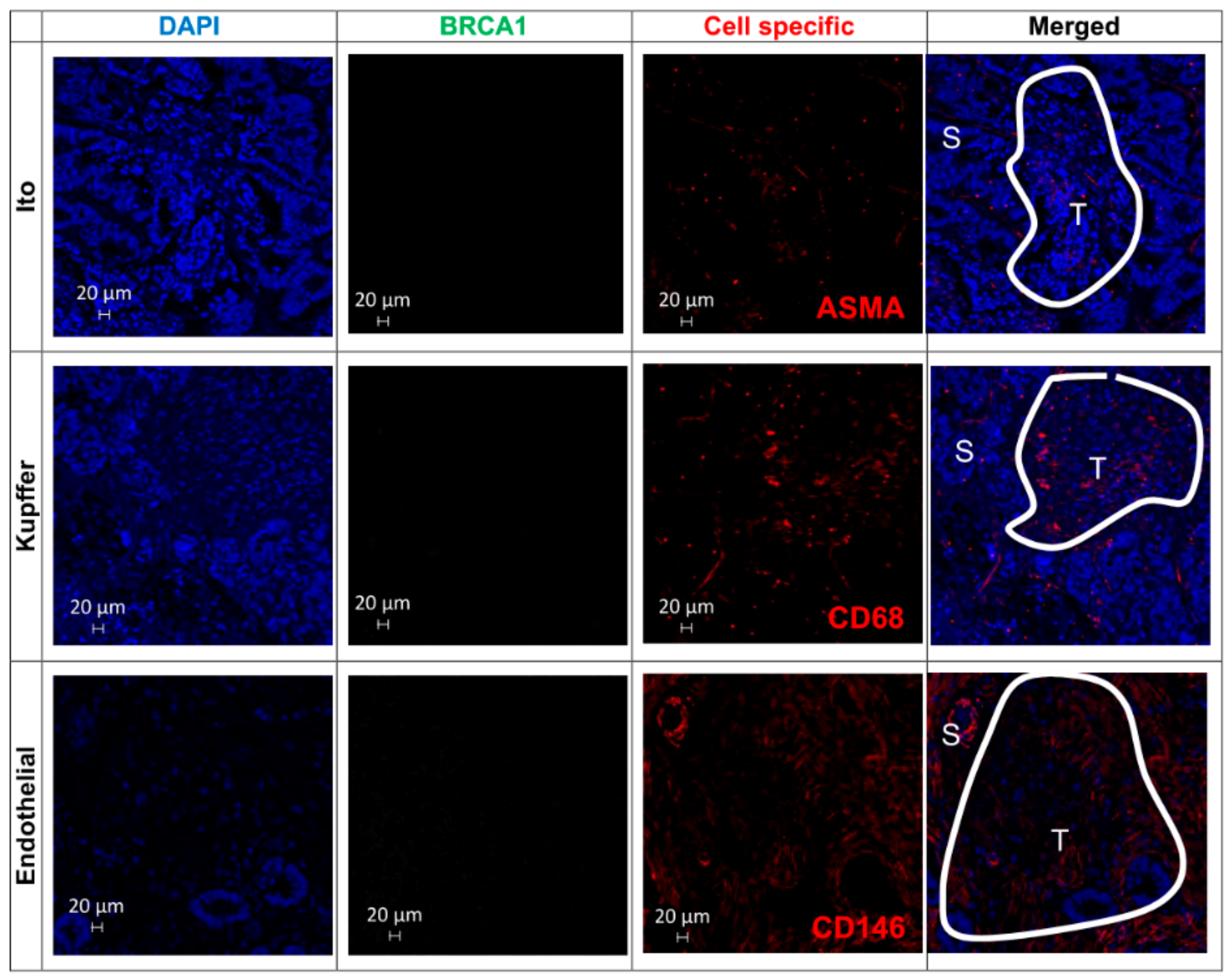
3.10. Immunohistochemical Analysis of TMAs from Human CRC Liver Metastases Reveals that BRCA1-Positive Samples Correlate with CRC Stage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Short Summary
Data Availability
Computer Code and Software
Abbreviations
| BASC | BRCA1-Associated genome Surveillance Complex |
| CRC | Colorectal Cancer |
| DEG | Differentially Expressed Gene |
| DEM | Differentially Expressed MicroRNA |
| DEP | Differentially Expressed Protein |
| EC | liver sinusoidal endothelial cell (E) from healthy/control (C) mice |
| ET | liver sinusoidal endothelial cell (E) from Tumor microenvironment (T) |
| IC | Ito cell (I) from healthy/control (C) mice |
| IT | Ito cell (I) from Tumor microenvironment (T) |
| KC | Kupffer cell (K) from healthy/control (C) mice |
| KT | Kupffer cell (K) from Tumor microenvironment (T) |
| miRNA | microRNA |
| TM | Tumor liver Metastasis |
| TME | Tumor Microenvironment |
| TMA | Tissue Microarray |
| TP | CRC Primary Tumor |
References
- World Cancer Research Fund; American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective. Continuous Update Project Expert Report. 2018. Available online: Dietandcancerreport.org (accessed on 21 January 2019).
- Langley, R.R.; Fidler, I.J. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr. Rev. 2007, 28, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J.; Poste, G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008, 9, 808. [Google Scholar] [CrossRef]
- Vidal-Vanaclocha, F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008, 1, 113–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaso, E.; Salado, C.; Egilegor, E.; Gutierrez, V.; Santisteban, A.; Sancho-Bru, P.; Friedman, S.L.; Vidal-Vanaclocha, F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 2003, 37, 674–685. [Google Scholar] [CrossRef]
- Badiola, I.; Olaso, E.; Crende, O.; Friedman, S.L.; Vidal-Vanaclocha, F. Discoidin domain receptor 2 deficiency predisposes hepatic tissue to colon carcinoma metastasis. Gut 2012, 61, 1465–1472. [Google Scholar] [CrossRef]
- Poschmann, G.; Seyfarth, K.; Besong Agbo, D.; Klafki, H.W.; Rozman, J.; Wurst, W.; Wiltfang, J.; Meyer, H.E.; Klingenspor, M.; Stühler, K. High-fat diet induced isoform changes of the Parkinson’s disease protein DJ-1. J. Proteome Res. 2014, 13, 2339–2351. [Google Scholar] [CrossRef]
- Gerovska, D.; Araúzo-Bravo, M.J. Computational analysis of single-cell transcriptomics data elucidates the stabilization of Oct4 expression in the E3.25 mouse preimplantation embryo. Sci. Rep. 2019, 9, 8930. [Google Scholar] [CrossRef] [Green Version]
- Holland, P.W.; Welsch, R.E. Robust regression using iteratively reweighted least-squares. communications in statistics: Theory and Methods. Commun. Stat. Theory Methods 1977, 6, 813–827. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods. 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cao, L.; Han, L.; Han, L.; Xu, Q.; Ma, Q. Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2/ERK/NF-κB axis. Int. J. Oncol. 2015, 46, 2613–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blander, G.; de Oliveira, R.M.; Conboy, C.M.; Haigis, M.; Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 2003, 278, 38966–38969. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Guo, Z.; Yao, K.; Miao, Y.; Liang, S.; Liu, F.; Wang, Y.; Zhang, Y. The transcriptional foundations of Sp110-mediated macrophage (RAW264.7) resistance to Mycobacterium tuberculosis H37Ra. Sci. Rep. 2016, 6, 22041. [Google Scholar] [CrossRef]
- Lyukmanova, E.N.; Shulepko, M.A.; Bychkov, M.L.; Shenkarev, Z.O.; Paramonov, A.S.; Chugunov, A.O.; Arseniev, A.S.; Dolgikh, D.A.; Kirpichnikov, M.P. Human SLURP-1 and SLURP-2 Proteins Acting on Nicotinic Acetylcholine Receptors Reduce Proliferation of Human Colorectal Adenocarcinoma HT-29 Cells. Acta Nat. 2014, 6, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Carlton, V.E.; Harris, B.Z.; Puffenberger, E.G.; Batta, A.K.; Knisely, A.S.; Robinson, D.L.; Strauss, K.A.; Shneider, B.L.; Lim, W.A.; Salen, G.; et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003, 34, 91–96. [Google Scholar] [CrossRef]
- Xiao, B.; Chen, D.; Luo, S.; Hao, W.; Jing, F.; Liu, T.; Wang, S.; Geng, Y.; Li, L.; Xu, W.; et al. Extracellular translationally controlled tumor protein promotes colorectal cancer invasion and metastasis through Cdc42/JNK/ MMP9 signaling. Oncotarget 2016, 7, 50057–50073. [Google Scholar] [CrossRef]
- Yamada, H.Y.; Gorbsky, G.J. Tumor suppressor candidate TSSC5 is regulated by UbcH6 and a novel ubiquitin ligase RING105. Oncogene 2006, 25, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, J.R.; Yamazaki, Y.; Palmer, R.H. Tumour-associated mutations of PA-TM-RING ubiquitin ligases RNF167/RNF13 identify the PA domain as a determinant for endosomal localization. Biochem. J. 2014, 459, 7–36. [Google Scholar] [CrossRef] [PubMed]
- Deroyer, C.; Rénert, A.F.; Merville, M.P.; Fillet, M. New role for EMD (emerin), a key inner nuclear membrane protein, as an enhancer of autophagosome formation in the C16-ceramide autophagy pathway. Autophagy 2014, 10, 1229–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Sharan, S.K. BRCA1 and MicroRNAs: Emerging networks and potential therapeutic targets. Mol. Cells 2012, 3, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, M.A.; Venturutti, L.; Huang, Y.W.; Schillaci, R.; Huang, T.H.; Elizalde, P.V. Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development. Breast Cancer Res. 2012, 14, R77. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.Z.; O’Connor, S.M.; van Holst, N.G.; Young, G.P.; James, R.J. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, S.T.; Chan, T.L.; Kwong, D.L.; Au, G.K.; et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Ciafrè, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2007, 65, 6029–6033. [Google Scholar] [CrossRef] [Green Version]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2004, 102, 3627–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, M.; Wilda, M.; Busch, K.; Viehmann, S.; Borkhardt, A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt’s lymphoma. Genes Chromosomes Cancer 2004, 39, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004, 64, 3087–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, M.V.; Visone, R.; Di Leva, G.; Donati, V.; Petrocca, F.; Casalini, P.; Taccioli, C.; Volinia, S.; Liu, C.G.; Alder, H.; et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007, 67, 8699–8707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Sayagués, J.M.; Corchete, L.A.; Gutiérrez, M.L.; Sarasquete, M.E.; Del Mar Abad, M.; Bengoechea, O.; Fermiñán, E.; Anduaga, M.F.; Del Carmen, S.; Iglesias, M.; et al. Genomic characterization of liver metastases from colorectal cancer patients. Oncotarget 2016, 7, 72908–72922. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zessin, A.S.; Glover, W.; Hsu, D.S. Activation of the mTOR pathway by Oxaliplatin in the treatment of colorectal cancer liver metastasis. PLoS ONE 2017, 12, e0169439. [Google Scholar] [CrossRef] [Green Version]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T.; et al. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Fenocchio, E.; Colombi, F.; Calella, M.G.; Filippi, R.; Depetris, I.; Chilà, G.; Lombardi, P.; Marino, D.; Cagnazzo, C.; Ferraris, R.; et al. Improvement of Metastatic Colorectal Cancer Patient Survival: Single Institution Experience. Cancers 2019, 11, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.F.; Howell, A.; Sartini, M.; Sotgia, F.; Lisanti, M.P. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1α, autophagy and ketone body production. Cell Cycle 2012, 11, 4167–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 modulates the immune status of the tumor microenvironment to facilitate metastatic colonization of colorectal cancer cells. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.; Zhao, X.; Qi, Y.; Zhang, Y.; Han, X.; Ding, Y.; Guan, J.; Ji, T.; Zhao, Y.; Nie, G. Reshaping prostate tumor microenvironment to suppress metastasis via cancer-associated fibroblast inactivation with peptide-assembly-based nanosystem. ACS Nano 2019, 13, 12357–12371. [Google Scholar] [CrossRef]
- Benedicto, A.; Romayor, I.; Arteta, B. CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol. Rep. 2018, 39, 2022–2030. [Google Scholar] [CrossRef]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in colorectal cancer liver metastases. Cancers 2019, 11, 633. [Google Scholar] [CrossRef] [Green Version]
- Valcárcel, M.; Carrascal, T.; Crende, O.; Vidal-Vanaclocha, F. IL-18 regulates melanoma VLA-4 integrin activation through a hierarchized sequence of inflammatory factors. J. Invest. Dermatol. 2014, 134, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Brodt, P. Role of the microenvironment in liver metastasis: From pre- to prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [Green Version]
- Badie, S.; Carlos, A.R.; Folio, C.; Okamoto, K.; Bouwman, P.; Jonkers, J.; Tarsounas, M. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J. 2015, 34, 410–424. [Google Scholar] [CrossRef]
- Bracci, M.; Ciarapica, V.; Zabaleta, M.E.; Tartaglione, M.F.; Pirozzi, S.; Giuliani, L.; Piva, F.; Valentino, M.; Ledda, C.; Rapisarda, V.; et al. BRCA1 and BRCA2 gene expression: Diurnal variability and influence of shift work. Cancers 2019, 11, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, D.; Abdouh, M.; Gao, Z.H.; Arena, V.; Arena, M.; Arena, G.O. Transfer of malignant trait to BRCA1 deficient human fibroblasts following exposure to serum of cancer patients. J. Exp. Clin. Cancer Res. 2016, 35, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arteta, B.; Lasuen, N.; Lopategi, A.; Sveinbjörnsson, B.; Smedsrød, B.; Vidal-Vanaclocha, F. Colon carcinoma cell interaction with liver sinusoidal endothelium inhibits organ-specific antitumor immunity through interleukin-1-induced mannose receptor in mice. Hepatology 2010, 51, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Wang, G.H.; Zhao, C.M.; Huang, Y.; Wang, W.; Zhang, S.; Wang, X. BRCA1 and BRCA2 expression patterns and prognostic significance in digestive system cancers. Hum. Pathol. 2018, 71, 135–144. [Google Scholar] [CrossRef]
- Etzold, A.; Galetzka, D.; Weis, E.; Bartsch, O.; Haaf, T.; Spix, C.; Itzel, T.; Schweiger, S.; Strand, D.; Strand, S.; et al. CAF-like state in primary skin fibroblasts with constitutional BRCA1 epimutation sheds new light on tumor suppressor deficiency-related changes in healthy tissue. Epigenetics 2016, 11, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, M.; Schinelli, S. Extracellular matrix alterations in metastatic processes. Int. J. Mol. Sci. 2019, 20, 4947. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wu, J.; Pan, C.; Wang, H.; Ying, X.; Zhou, Y.; Yu, H.; Zuo, Y.; Pan, Z.; Liu, R.Y.; et al. Genetic and epigenetic down-regulation of microRNA-212 promotes colorectal tumor metastasis via dysregulation of MnSOD. Gastroenterology 2013, 145, 426–436. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Activation Sequence | R | N | Genes |
|---|---|---|---|
| Endothelial→Ito→Kupffer | ↑ | 34 | 3110047P20Rik, AW060742, A_55_P1964173, A_55_P2091525, Abca17, Armcx3, Atp8a1, B930078G14Rik, Cd300a, Elavl4, Fam160a2, Fdxacb1, Fnbp1, Gm10914, Golga7b, Med13, Nhlh1, Npcd, Rdh1, Slc1a4, TC1648127, Zfp133-ps, chr10:69842010-69842465_F, chr11:53574182-53574644_R, chr14:78214602-78225827_F, chr17:17462450-17488725_R, chr1:138442536-138521080_F, chr1:182686731-182709331_F, chr4:131905739-131910814_R, chr5:74340084-74346709_R, chr6:136440100-136450050_F, chr6:146493273-146493932_R,chr8:35143801-35144482_F, chrX:120300373-120302404_F |
| ↓ | 0 | ||
| Endothelial→Kupffer→Ito | ↑ | 16 | 1300017J02Rik, Aadac, Afm, Ahsg, Apof, C8a, C8b, Cyp2a12, Cyp2d10, Cyp2d26, Habp2, Itih, Itih2, Serpinc1, Serpind1, Ugt2b36 |
| ↓ | 11 | Apln, Edn1 (10 probes) | |
| Ito→Endothelial→Kupffer | ↑ | 0 | |
| ↓ | 1 | Tm4sf4 |
| Cell Activation Sequence | R | N | Genes |
|---|---|---|---|
| Endothelial→Ito→Kupffer | ↑ | 4 | Igf2bp, Prss46, Slc6a4, Tnfsf4 |
| ↓ | 0 | ||
| Endothelial→Kupffer→Ito | ↑ | 0 | |
| ↓ | 0 | ||
| Ito→Endothelial→Kupffer | ↑ | 6 | Apob, Beta-s, Clec4b1, Hba-a2, Hbb-b1, Hbb-b2 |
| ↓ | 1 | Apol10b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerovska, D.; Larrinaga, G.; Solano-Iturri, J.D.; Márquez, J.; García Gallastegi, P.; Khatib, A.-M.; Poschmann, G.; Stühler, K.; Armesto, M.; Lawrie, C.H.; et al. An Integrative Omics Approach Reveals Involvement of BRCA1 in Hepatic Metastatic Progression of Colorectal Cancer. Cancers 2020, 12, 2380. https://doi.org/10.3390/cancers12092380
Gerovska D, Larrinaga G, Solano-Iturri JD, Márquez J, García Gallastegi P, Khatib A-M, Poschmann G, Stühler K, Armesto M, Lawrie CH, et al. An Integrative Omics Approach Reveals Involvement of BRCA1 in Hepatic Metastatic Progression of Colorectal Cancer. Cancers. 2020; 12(9):2380. https://doi.org/10.3390/cancers12092380
Chicago/Turabian StyleGerovska, Daniela, Gorka Larrinaga, Jon Danel Solano-Iturri, Joana Márquez, Patricia García Gallastegi, Abdel-Majid Khatib, Gereon Poschmann, Kai Stühler, María Armesto, Charles H. Lawrie, and et al. 2020. "An Integrative Omics Approach Reveals Involvement of BRCA1 in Hepatic Metastatic Progression of Colorectal Cancer" Cancers 12, no. 9: 2380. https://doi.org/10.3390/cancers12092380
APA StyleGerovska, D., Larrinaga, G., Solano-Iturri, J. D., Márquez, J., García Gallastegi, P., Khatib, A.-M., Poschmann, G., Stühler, K., Armesto, M., Lawrie, C. H., Badiola, I., & Araúzo-Bravo, M. J. (2020). An Integrative Omics Approach Reveals Involvement of BRCA1 in Hepatic Metastatic Progression of Colorectal Cancer. Cancers, 12(9), 2380. https://doi.org/10.3390/cancers12092380