Systems Biology Approaches Reveal Potential Phenotype-Modifier Genes in Neurofibromatosis Type 1

 , ,
, ,
Abstract
:1. Introduction
2. Results
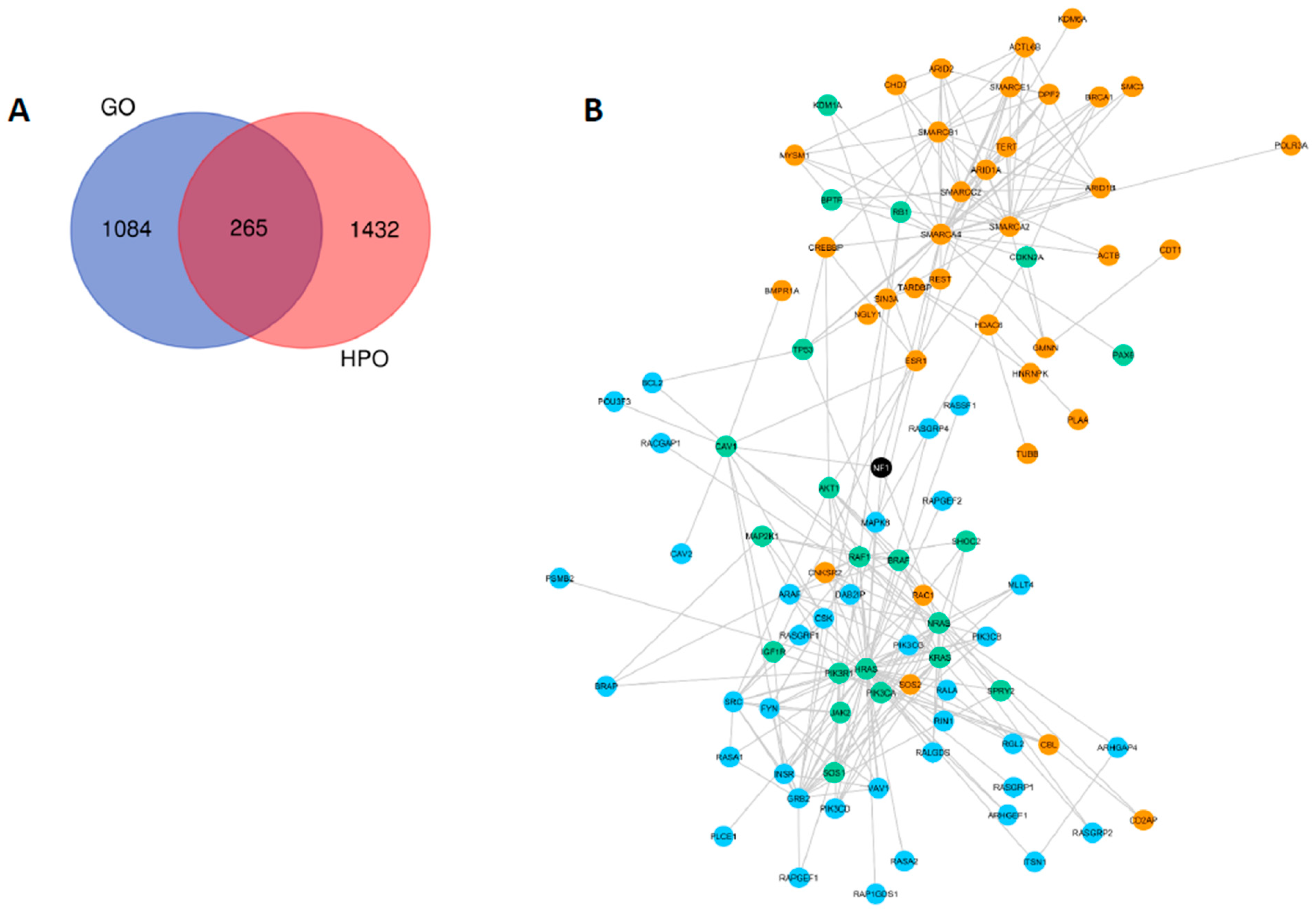
2.1. Gene and Phenotype Ontologies Analyses
2.2. Network Statistics
2.3. Forward and Reverse Genetics Strategies
2.4. Differential Gene Expression Networks
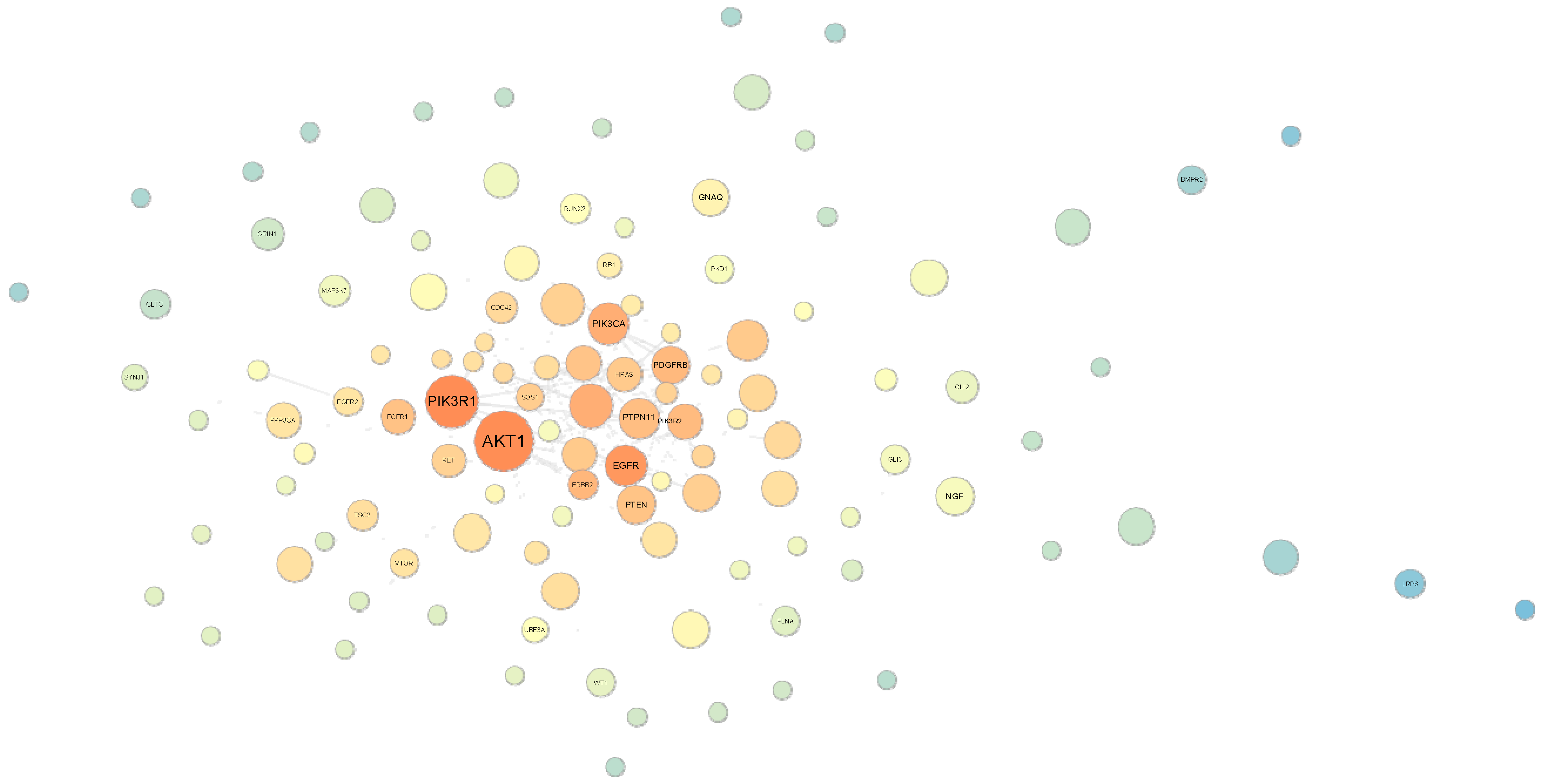
2.5. Systems Biology Approaches Reveal 10 NF1 Phenotype-Modifier Candidate Genes
2.6. Random Walk Analysis
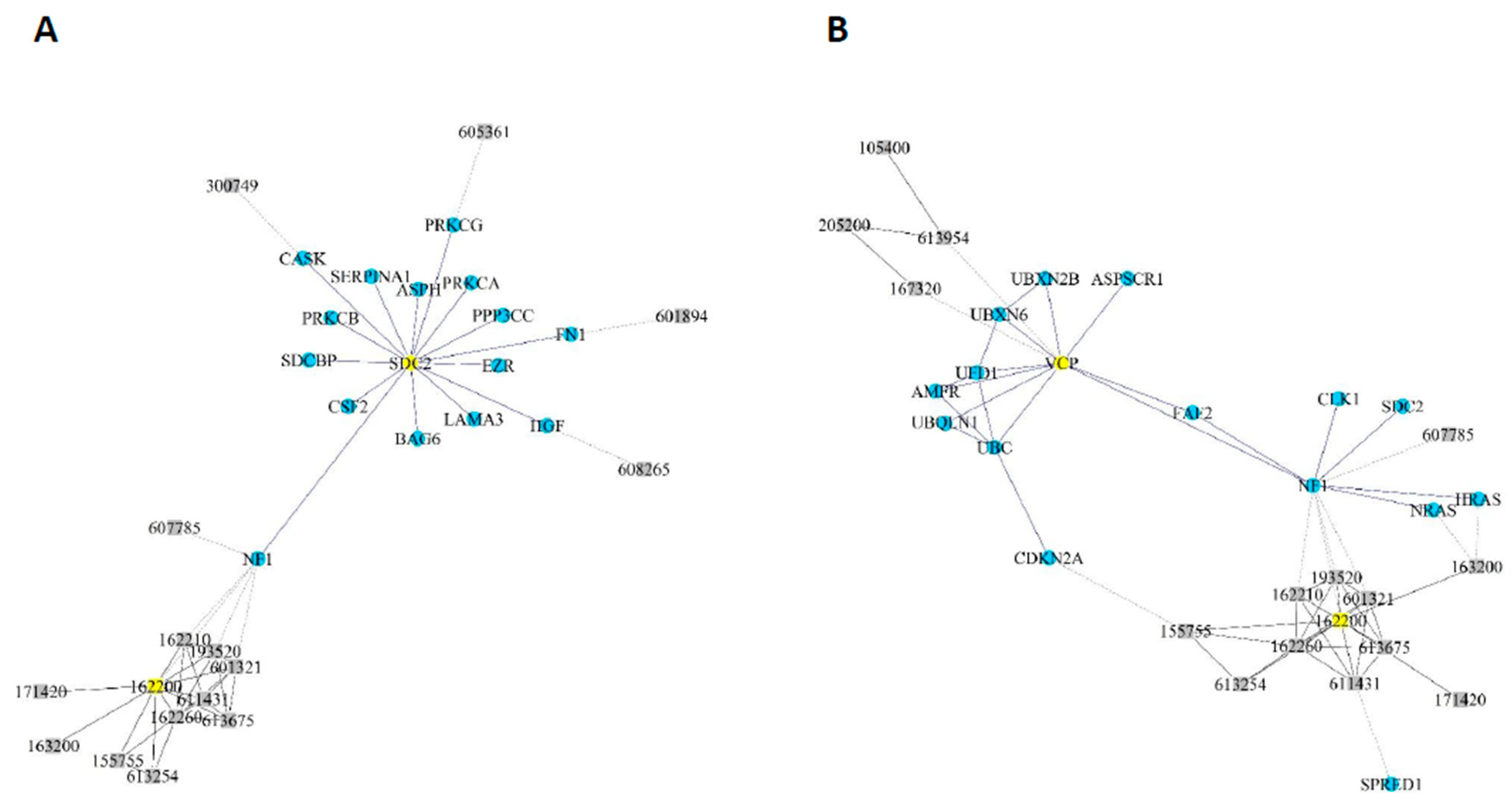
2.7. Literature Review and Genomic Databases Evaluation
3. Discussion
4. Materials and Methods
4.1. Selection of NF1 Ontologies
4.2. Systems Biology Analysis
4.3. Gene Expression Evaluation
4.4. Random Walk Analysis
4.5. Database Research
4.6. Variant Datasets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, D.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.; Lalloo, F.; Evans, D.G. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. Part A 2010, 152, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2009, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Evans, D.G.; Baser, M.E.; McGaughran, J.M.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Seminog, O.; Goldacre, M.J. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: Population-based record-linkage study. Br. J. Cancer 2012, 108, 193–198. [Google Scholar] [CrossRef]
- Carey, J.C.; Baty, B.J.; Johnson, J.P.; Morrison, T.; Skolnick, M.; Kivlin, J. The genetic aspects of neurofibromatosis. Ann. N. Y. Acad. Sci. 1986, 486, 45–56. [Google Scholar] [CrossRef]
- Mensink, K.A.; Ketterling, R.P.; Flynn, H.C.; Knudson, R.A.; Lindor, N.M.; Heese, B.A.; Spinner, R.J.; Babovic-Vuksanovic, D. Connective tissue dysplasia in five new patients with NF1 microdeletions: Further expansion of phenotype and review of the literature. J. Med. Genet. 2005, 43, e8. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated Risk for MPNST in NF1 Microdeletion Patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Descheemaeker, M.; Roelandts, K.; De Raedt, T.; Brems, H.; Fryns, J.; Legius, E. Intelligence in individuals with a neurofibromatosis type 1 microdeletion. Am. J. Med. Genet. 2004, 131, 325–326. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp Inframe deletion in Exon 17 of the NF1 Gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 Genotype-Phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2014, 23, 1068–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.-C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype correlation in NF1: Evidence for a more severe Phenotype associated with Missense mutations affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef]
- Mautner, V.-F.; Kluwe, L.; Friedrich, R.; Roehl, A.C.; Bammert, S.; Högel, J.; Cooper, D.N.; Kehrer-Sawatzki, H.; Spöri, H. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyaya, S.A.; McGee, R.B.; Wilky, B.A.; Broniscer, A. Malignant progression of a peripheral nerve sheath tumor in the setting of rhabdoid tumor predisposition syndrome. Pediatr. Blood Cancer 2018, 65, e27030. [Google Scholar] [CrossRef] [PubMed]
- Grisart, B.; Rack, K.; Vidrequin, S.; Hilbert, P.; Deltenre, P.; Verellen-Dumoulin, C.; Destrée, A.; Destr, A. NF1 microduplication first clinical report: Association with mild mental retardation, early onset of baldness and dental enamel hypoplasia? Eur. J. Hum. Genet. 2008, 16, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Pasmant, E.; Laurendeau, I.; Parfait, B.; Barbarot, S.; Guillot, B.; Combemale, P.; Ferkal, S.; Vidaud, M.; Aubourg, P.; et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum. Mol. Genet. 2009, 18, 2768–2778. [Google Scholar] [CrossRef]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- Pasmant, E.; Vidaud, M.; Vidaud, D.; Wolkenstein, P. Neurofibromatosis type 1: From genotype to phenotype. J. Med. Genet. 2012, 49, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Pemov, A.; Sung, H.; Hyland, P.L.; Sloan, J.L.; Ruppert, S.L.; Baldwin, A.M.; Boland, J.F.; Bass, S.E.; Lee, H.J.; Jones, K.M.; et al. Genetic modifiers of neurofibromatosis type 1-associated café-au-lait macule count identified using multi-platform analysis. PLoS Genet. 2014, 10, e1004575. [Google Scholar] [CrossRef] [Green Version]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.-J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [Green Version]
- Tritto, V.; Ferrari, L.; Esposito, S.; Zuccotti, P.; Bianchessi, D.; Natacci, F.; Saletti, V.; Eoli, M.; Riva, P. Non-coding RNA and tumor development in neurofibromatosis Type 1: ANRIL Rs2151280 is associated with optic glioma development and a mild phenotype in neurofibromatosis Type 1 Patients. Genes 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasmant, E.; Sabbagh, A.; Masliah-Planchon, J.; Ortonne, N.; Laurendeau, I.; Melin, L.; Ferkal, S.; Hernandez, L.; Leroy, K.; Valeyrie-Allanore, L.; et al. Role of Noncoding RNA ANRIL in Genesis of Plexiform Neurofibromas in Neurofibromatosis Type 1. J. Natl. Cancer Inst. 2011, 103, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Muram-Zborovski, T.M.; Stevenson, D.A.; Viskochil, D.H.; Dries, D.C.; Wilson, A.; Mao, R. SPRED 1 mutations in a neurofibromatosis clinic. J. Child Neurol. 2010, 25, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Xu, S.; Liu, R.; Shi, T.; Li, X.; Li, X.; Chen, G.; Liu, H.; Zhou, Q.; Chen, J. The investigation for potential modifier genes in patients with neurofibromatosis type 1 based on next-generation sequencing. OncoTargets Ther. 2018, 11, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharafi, P.; Ayter, S. Possible modifier genes in the variation of neurofibromatosis type 1 clinical phenotypes. J. Neurogenet. 2018, 32, 65–77. [Google Scholar] [CrossRef]
- Le Novère, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, T.W.; Dupont, Á.D.V.; Rengel, B.D.; Sgarioni, E.; Gomes, J.D.A.; Fraga, L.R.; Schuler-Faccini, L.; Vianna, F.S.L. Assembling systems biology, embryo development and teratogenesis: What do we know so far and where to go next? Reprod. Toxicol. 2019, 88, 67–75. [Google Scholar] [CrossRef]
- Verster, J.C.; Tzschentke, T.M.; O’Malley, K.; Colpaert, F.C.; Ellenbroek, B.; McAllister-Williams, R.H.; Liepert, J.; Hillard, C.J.; Preskorn, S.; Dahmen, M.M.; et al. Forward Genetics/Reverse Genetics. In Encyclopedia of Psychopharmacology; Springer Science and Business Media LLC: Berlin, Germany, 2010; p. 544. [Google Scholar]
- Köhler, S.; Carmody, L.C.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2018, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, Q.; Katsevich, E.; Sabatti, C. Exploratory gene ontology analysis with interactive visualization. Sci. Rep. 2019, 9, 7793. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Boil. 2007, 3, e59. [Google Scholar] [CrossRef]
- Valdeolivas, A.; Tichit, L.; Navarro, C.; Perrin, S.; Odelin, G.; Levy, N.; Cau, P.; Remy, E.; Baudot, A. Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics 2018, 35, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warrington, N.M.; Sun, T.; Luo, J.; McKinstry, R.C.; Parkin, P.C.; Ganzhorn, S.; Spoljaric, D.; Albers, A.C.; Merkelson, A.; Stewart, U.R.; et al. The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res. 2014, 75, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Mo, W.; Chen, J.; Patel, A.; Zhang, L.; Chau, V.; Li, Y.; Cho, W.; Lim, K.; Xu, J.; Lazar, A.J.F.; et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell 2013, 152, 1077–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahuau, M.; Pelet, A.; Vidaud, M.; Lamireau, T.; Le Bail, B.; Munnich, A.; Vidaud, M.; Lyonnet, S.; Lacombe, D. GDNF as a candidate modifier in a type 1 neurofibromatosis (NF1) enteric phenotype. J. Med. Genet. 2001, 38, 638–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Thayanithy, V.; West, R.B.; Lee, C.-H.; Beck, A.H.; Zhu, S.; Downs-Kelly, E.; Montgomery, K.; Goldblum, J.R.; Hogendoorn, P.C.; et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J. Pathol. 2010, 220, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, S.; Kunisada, T.; Morimoto, Y.; Yoshida, A.; Sasaki, T.; Ito, S.; Ouchida, M.; Sugihara, S.; Shimizu, K.; Ozaki, T. MicroRNA-21 correlates with tumorigenesis in malignant peripheral nerve sheath tumor (MPNST) via programmed cell death protein 4 (PDCD4). J. Cancer Res. Clin. Oncol. 2012, 138, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Gong, M.; Ma, J.; Li, M.; Zhou, M.; Hock, J.; Yu, X. MicroRNA-204 critically regulates carcinogenesis in malignant peripheral nerve sheath tumors. Neuro-oncology 2012, 14, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef]
- Wang, J.H. Succinate Dehydrogenase subunit B (SDHB) is expressed in neurofibromatosis 1-associated Gastrointestinal Stromal Tumors (Gists): Implications for the SDHB expression based classification of Gists. J. Cancer 2011, 2, 90. [Google Scholar] [CrossRef]
- Nadim, W.D.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.-L.; Zajdel, P.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. Physical interaction between neurofibromin and serotonin 5-HT6receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef] [Green Version]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.-P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1–like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.E.; Grimstead, J.W.; Sedani, A.; Baird, D.M.; Upadhyaya, M. Telomere erosion in NF1 tumorigenesis. Oncotarget 2017, 8, 40132–40139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahit, K.T.H.; Tarailo-Graovac, M. Genetic modifiers and rare mendelian disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Fuente, A. From ‘differential expression’ to ‘differential networking’—Identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Saavedra, E.; Gallardo-Pérez, J.C.; Rumjanek, F.D.; Rodríguez-Enríquez, S. Understanding the cancer cell phenotype beyond the limitations of current omics analyses. FEBS J. 2015, 283, 54–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczinger, M.; Kemény, L. Novel Factors in the Pathogenesis of Psoriasis and Potential Drug Candidates Are Found with Systems Biology Approach. PLoS ONE 2013, 8, e80751. [Google Scholar] [CrossRef] [Green Version]
- Carapito, R.; Carapito, C.; Morlon, A.; Paul, N.; Jacome, A.S.V.; Alsaleh, G.; Rolli, V.; Tahar, O.; Aouadi, I.; Rompais, M.; et al. Multi-OMICS analyses unveil STAT1 as a potential modifier gene in mevalonate kinase deficiency. Ann. Rheum. Dis. 2018, 77, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Loviglio, M.N.; Beck, C.R.; White, J.; Leleu, M.; Harel, T.; Guex, N.; Niknejad, A.; Bi, W.; Chen, E.S.; Crespo, I.; et al. Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med. 2016, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Ghatge, M.; Nair, J.; Sharma, A.; Vangala, R.K. Integrative gene ontology and network analysis of coronary artery disease associated genes suggests potential role of ErbB pathway gene EGFR. Mol. Med. Rep. 2018, 17, 4253–4264. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, J.; Zhang, X.; Wang, X.; Jiang, L.; Gu, X. Identification of AIDS-associated Kaposi Sarcoma: A functional genomics approach. Front. Genet. 2020, 10, 1376. [Google Scholar] [CrossRef]
- Worley, K.; Rico-Varela, J.; Ho, D.; Wan, L.Q. Teratogen screening with human pluripotent stem cells. Integr. Boil. 2018, 10, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, B.C.; Wu, J.; Miller, S.J.; Monk, K.R.; Shamekh, R.; Rizvi, T.A.; Decourten-Myers, G.; Vogel, K.S.; DeClue, J.E.; Ratner, N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell 2005, 7, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, W.; Williams, J.P.; Ratner, N. EGFR-Stat3 signalling in nerve glial cells modifies neurofibroma initiation. Oncogene 2016, 36, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Orloff, M.S.; He, X.; Peterson, C.; Chen, F.; Chen, J.-L.; Mester, J.L.; Eng, C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2013, 92, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhunapantula, S.V.; Mosca, P.J.; Robertson, G.P. The Akt signaling pathway. Cancer Boil. Ther. 2011, 12, 1032–1049. [Google Scholar] [CrossRef] [Green Version]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 inactivation in Schwann cells produces a severe Phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Gregorian, C.; Nakashima, J.; Dry, S.M.; Nghiemphu, P.L.; Smith, K.B.; Ao, Y.; Dang, J.; Lawson, G.; Mellinghoff, I.K.; Mischel, P.S.; et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 19479–19484. [Google Scholar] [CrossRef] [Green Version]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F.R.; et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Wu-Chou, Y.-H.; Hung, T.-C.; Lin, Y.-T.; Cheng, H.-W.; Lin, J.-L.; Lin, C.-H.; Yu, C.-C.; Chen, P.; Yeh, T.-H.; Chen, Y.-R. Genetic diagnosis of neurofibromatosis type 1: Targeted next- generation sequencing with Multiple Ligation-Dependent Probe Amplification analysis. J. Biomed. Sci. 2018, 25, 72. [Google Scholar] [CrossRef]
- Manetti, F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med. Res. Rev. 2011, 32, 968–998. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.; Olson, M.F. LIM kinases: Function, regulation and association with human disease. J. Mol. Med. 2007, 85, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Marwaha, S.; Rutkowski, J.L.; Tennekoon, G.I.; Phillips, P.C.; Field, J. A role for Pak protein kinases in Schwann cell transformation. Proc. Natl. Acad. Sci. USA 1998, 95, 5139–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Chen, Z.; Ambrose, D.; Liu, J.; Gibbs, J.B.; Chernoff, J.; Field, J. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol. Cell. Boil. 1997, 17, 4454–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Arnaud, O.; Le Loarer, F.; Tirode, F. BAFfling pathologies: Alterations of BAF complexes in cancer. Cancer Lett. 2018, 419, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Jo, V.Y.; Fletcher, C.D.M. SMARCB1/INI1 loss in Epithelioid Schwannoma. Am. J. Surg. Pathol. 2017, 41, 1013–1022. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Weihl, C.C. Another VCP interactor: NF is enough. J. Clin. Investig. 2011, 121, 4627–4630. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-F.; Shih, Y.-T.; Chen, C.-Y.; Chao, H.-W.; Lee, M.-J.; Hsueh, Y.-P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.-P.; Yang, F.-C.; Kharazia, V.; Naisbitt, S.; Cohen, A.R.; Weinberg, R.J.; Sheng, M. Direct interaction of CASK/LIN-2 and Syndecan Heparan sulfate Proteoglycan and their overlapping distribution in neuronal synapses. J. Cell Boil. 1998, 142, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anney, R.J. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 2017, 8, 21. [Google Scholar] [CrossRef]
- Plasschaert, E.; Descheemaeker, M.-J.; Van Eylen, L.; Noens, I.; Steyaert, J.; Legius, E. Prevalence of Autism Spectrum Disorder symptoms in children with neurofibromatosis type 1. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 168, 72–80. [Google Scholar] [CrossRef]
- Zhang, W.; Lei, X.; Bian, C. Identifying cancer genes by combining two-rounds RWR based on multiple biological data. BMC Bioinform. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Yoo, S.; Nam, H.; Lee, D.-J. Phenotype-oriented network analysis for discovering pharmacological effects of natural compounds. Sci. Rep. 2018, 8, 11667. [Google Scholar] [CrossRef] [Green Version]
- Rieley, M.B.; Stevenson, D.A.; Viskochil, D.H.; Tinkle, B.T.; Martin, L.J.; Schorry, E. Variable expression of neurofibromatosis 1 in monozygotic twins. Am. J. Med. Genet. Part A 2011, 155, 478–485. [Google Scholar] [CrossRef]
- Szudek, J.; Joe, H.; Friedman, J. Analysis of intrafamilial phenotypic variation in neurofibromatosis 1 (NF1). Genet. Epidemiol. 2002, 23, 150–164. [Google Scholar] [CrossRef]
- Malone, J.R.; Stevens, R.; Jupp, S.; Hancocks, T.; Parkinson, H.; Brooksbank, C. Ten Simple Rules for Selecting a Bio-ontology. PLoS Comput. Boil. 2016, 12, e1004743. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (OMIM) | Aliases | Cytogenetic Location | Summary | PINOT 1 | STRING 2 | BioGrid | Human Interactome | HPO | GO | Phenome Scape | Direct Strategy | Forward Strategy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AKT1 (*164730) | AKT, CWS6, PKB, PKB-ALPHA, PRKBA, RAC, RAC-ALPHA | 14q32.33 | Serine/threonine kinase - development of the human nervous system; mediator of growth factor-induced neuronal survival; can inactivate components of apoptosis | X | X | X | X | X | ||||
| BRAF (164757) | NS7; B-raf; BRAF1; RAFB1; B-RAF1 | 7q34 | Serine/threonine kinase - role in regulating the MAP kinase/ERK signaling pathway | X | X | |||||||
| EGFR (131550) | ERBB, ERBB1, HER1, NISBD2, PIG61, mENA | 7p11.2 | Cell surface protein - acts as a receptor for members of the epidermal growth factor family which induces cell proliferation | X | X | X | X | |||||
| LIMK1 (601329) | LIMK; LIMK-1 | 7q11.23 | Serine/threonine kinase - regulates actin polymerization; it is ubiquitously expressed during development; associated with cytoskeletal structure | X | X | |||||||
| PAK1 (602590) | IDDMSSD; p65-PAK; PAKalpha; alpha-PAK | 11q13.5-q14.1 | Serine/threonine kinase - cytoskeleton reorganization and nuclear signaling; regulates cell motility and morphology; essential for the RAS-induced malignant transformation | X | X | |||||||
| PTEN (601728) | BZS; DEC; CWS1; GLM2; MHAM; TEP1; MMAC1; PTEN1; PTENbeta | 10q23.31 | Phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase that functions as a tumor suppressor | X | X | X | X | X | X | |||
| RAF1 (164760) | NS5; CRAF; Raf-1; c-Raf; CMD1NN | 3p25.2 | MAP3 kinase - involved in the cell division cycle, apoptosis, cell differentiation and cell migration | X | X | X | X | X | ||||
| SDC2 (142460) | HSPG; CD362; HSPG1; SYND2 | 8q22.1 | Syndecan proteoglycan protein - mediates cell binding, cell signaling, and cytoskeletal organization | X | X | X | X | X | ||||
| SMARCA4 (603254) | BRG1; CSS4; SNF2; SWI2; MRD16; RTPS2; BAF190; SNF2L4; SNF2LB; hSNF2b; BAF190A; SNF2-beta | 19p13.2 | Part of the large ATP-dependent chromatin remodeling complex required for transcriptional activation of genes normally repressed by chromatin | X | X | X | X | |||||
| VCP (601023) | p97; TERA; CDC48 | 9p13.3 | Plays a role in protein degradation, intracellular membrane fusion, DNA repair and replication, regulation of the cell cycle, and activation of the NF-kappa B pathway | X | X | X | X | X | X |
| GENE | All | Missense | Synonymous | Splice Site | Frameshift | Inframe Del/Ins | Intronic | Nonsense | Stop Lost | Start Lost | 5′UTR | 3′UTR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AKT1 | 932 | 166 (17.81%) | 155 (16.63%) | 80 (8.58%) | 3 (0.32%) | 4 (0.43%) | 435 (46.67%) | 1 (0.11%) | 0 | 0 | 36 (3.86%) | 52 (5.58%) |
| BRAF | 1073 | 230 (21.44%) | 170 (15.84%) | 56 (5.22%) | 1 (0.09%) | 8 (0.75%) | 561 (52.28%) | 2 (0.19%) | 1 (0.09%) | 0 | 13 (1.21%) | 31 (2.89%) |
| EGFR | 2000 | 682 (34.10%) | 387 (19.35%) | 117 (5.85%) | 15 (0.75%) | 2 (0.10%) | 867 (43.35%) | 16 (0.80%) | 1 (0.05%) | 0 | 9 (0.45%) | 104 (5.20%) |
| LIMK1 | 1133 | 322 (28.42%) | 214 (18.89%) | 58 (5.12%) | 5 (0.44%) | 1 (0.09%) | 469 (41.39%) | 3 (0.26%) | 1 (0.09%) | 0 | 44 (3.88%) | 16 (1.41%) |
| PAK1 | 774 | 128 (16.54%) | 121 (15.63%) | 58 (7.49%) | 4 (0.52%) | 6 (0.78%) | 401 (51.81%) | 5 (0.65%) | 1 (0.13%) | 0 | 8 (1.03%) | 42 (5.43%) |
| PTEN | 456 | 83 (18.20%) | 77 (16.89%) | 17 (3.73%) | 5 (1.10%) | 0 | 223 (48.90%) | 5 (1.10%) | 0 | 0 | 28 (6.14%) | 18 (3.95%) |
| RAF1 | 1005 | 264 (26.27%) | 145 (14.43%) | 65 (6.47%) | 4 (0.40%) | 2 (0.20%) | 490 (48.76%) | 7 (0.70%) | 0 | 2 (0.20%) | 8 (0.80%) | 18 (1.79%) |
| SDC2 | 335 | 102 (30.45%) | 52 (15.52%) | 18 (5.37%) | 5 (1.49%) | 3 (0.90%) | 114 (34.03%) | 0 | 0 | 5 (1.49%) | 22 (6.57%) | 14 (4.18%) |
| SMARCA4 | 2575 | 473 (18.37%) | 551 (21.40%) | 164 (6.37%) | 4 (0.16%) | 22 (0.85%) | 1318 (51.18%) | 0 | 1 (0.04%) | 0 | 9 (0.35%) | 33 (1.28%) |
| VCP | 928 | 135 (14.55%) | 191 (20.58%) | 72 (7.76%) | 1 (0.11%) | 1 (0.11%) | 488 (52.59%) | 0 | 0 | 0 | 20 (2.16%) | 20 (2.16%) |
| GENE | Classification * | Related Syndromes *** | Other Relevant Reported Conditions | ||||
|---|---|---|---|---|---|---|---|
| All ** | B/LB | P/LP | CI | VUS | |||
| AKT1 | 182 | 94 | 8 | 4 | 76 | Cowden, Proteus | E17K variant was associated with 22 conditions, including breast cancer |
| BRAF | 334 | 125 | 80 | 12 | 117 | Cardiofaciocutaneous, Dandy-Walker malformation, LEOPARD, PHACE, Noonan | Astrocytoma, glioma |
| EGFR | 199 | 54 | 4 | 7 | 134 | Cowden, not otherwise specified (NOS) hereditary cancer | Cerebral arteriovenous malformation, inflammatory skin and bowel disease |
| LIMK1 | 36 | 36 | 0 | 0 | 0 | - | - |
| PAK1 | 6 | 2 | 3 | - | 1 | - | intellectual developmental disorder with macrocephaly, seizures, and speech delay |
| PTEN | 1567 | 367 | 510 | 22 | 668 | Bannayan-Riley-Ruvalcaba, Cowden, Hereditary breast and ovarian cancer, NOS Hereditary cancer-predisposing, Proteus-like | Macrocephaly/autism, Phophatase and Tensin (PTEN) Homolog hamartoma tumor |
| RAF1 | 412 | 155 | 43 | 17 | 197 | LEOPARD, Noonan | Chordoma, retinoblastoma, and |
| SDC2 | - | - | - | - | - | - | leri pleonosteosis are reported in patients carrying the copy number gain of 8q22.1, which includes SDC2 |
| SMARCA4 | 2310 | 980 | 95 | 81 | 1154 | Coffin-Siris, NOS Hereditary cancer-predisposing, Rhabdoid tumor predisposition | Craniopharyngioma, intellectual deficiency, medulloblastoma, neurodevelopmental disorder, neuroblastoma |
| VCP | 170 | 64 | 18 | 10 | 78 | - | Amyotrophic lateral sclerosis, paget disease, Charcot-Marie-Thoth disease |
| Genes/Proteins | Consequence | Methodology Aspects | Reference |
|---|---|---|---|
| ADCY8 | Genetic polymorphisms in ADCY8 are correlated with glioma risk in NF1 in a sex-specific manner, elevating risk in females while reducing risk in males | - Genotyping of NF1 patients using Affymetrix whole-genome human SNP array - Primary astrocyte cultures from NF1-CKO mice and treatment with dideoxyadenosine to induce ADCY inhibition - cAMP regulator expression with qPCR and ELISA | Warrington et al. 2015 [33] |
| ANRIL allele T of SNP rs2151280 | Higher number of plexiform neurofibromas; rs2151280 reduced ANRIL transcript levels | - High-resolution array comparative genomic hybridization (aCGH) of PNFs from NF1 patients | Pasmant et al. 2011 [22] |
| ATP6V0B SNP rs7161 DPH2 SNP rs4660761 MSH6 SNP rs1800934 | ATP6V0B is associated with melanosome biology rs7161 and rs4660761 associated with café-au-lait macule (CALM) count; rs1800934 associated with development an NF1-like phenotype | - Lymphoblastoid cell lines with NF1-associated phenotypes - Gene expression (microarray and qPCR) - Sequencing of genes with incresased expression in patients with high count CALM - Meta-analysis | Pemov et al. 2014 [19] |
| CRLF3, ADAP2, RNF135, UTP6, SUZ12, OMG, LRRC37B, EVI2A, EVI2B, RAB11FIP4, RAB11FIP3, TEFM, ATAD5, CORPS, NF1 large 17q11 deletions encompassing the entire NF1 locus and neighboring genes | Dysmorphic features, learning disabilities, cardiovascular malformations, childhood overgrowth, a higher tumor burden and earlier onset of benign neurofibromas, and probably, a higher incidence of malignant peripheral nerve sheath tumors (MPSTs) | -MLPA, breakpoint-spanning PCR and FISH in NF1 deletion patients | Mautner et al. 2010 [13] |
| CXCR4 and its ligand, CXCL12 | Highly expressed in mouse models of NF1-deficient MPNSTs, but not in normal precursor cells; Suppression of CXCR4 activity decreases MPNST cell growth in culture and inhibits tumorigenesis in allografts and in spontaneous genetic mouse models of MPNST; Demonstrated conservation of these activated molecular pathways in human MPNSTs | - NF1-deficient skin-derived precursor models (SKPs) and gene expressuion microarray (normal SKPs; pretumorigenic SKPs with either Nf1 deletion or Nf1 and p53 deletion) - qPCR, westerblot and IHC of CXCR4 and CXCL12 - CXCR4 shRNA for knockdown in SKP MPNST cells - Tissue microarray from plexiform neurofibromas in NF1 patients harboring MPNSTs and MPNSTs samples from NF1 and sporadic patients - CXCR4 cDNAs sequencing | Mo et al. 2013 [34] |
| GDNF R93W germiline variant and maternally inherited NF1 mutation | Congenital megacolon development | - Investigation of a family carrying variants in GDNF and NRTN genes with cutaneous manifestations of NF1 and megacolon - Haploinsufficient animal models for Nf1 and Trp53 that developed MPNSTs | Bahuau et al. 2001 [35] |
| miR-34a | Down-regulation of miR-34a founded in most MPNSTs compared to neurofibromas; The p53 inactivation and subsequent loss of expression of miR-34a may contribute to MPNST development | - Microarray of MPNSTs, neurofibromas, Schwannonas, and synovial sarcomas - MPNST cell lines to check for miR-34a and other p53-dependent miRNAs by qRT-PCR after overexpressing wild-type p53 | Subramanian et al. 2010 [36] |
| miR-21 | Important in MPNST tumorigenesis and progression through its target, PDCD4 | - Global miRNA expression profiling of MPNSTs and neurofibromas - qPCR of differentially expressed miRNAs in MPNSTs, 11 NFs, and 5 normal nerves and MPNST cell lines - Knockdown of miR-21 in MPNST cells | Itani et al. 2012 [37] |
| miR-204 | Down-regulation of miR-204 contributes to development and tumor progression of MPNSTs | - Global miRNA expression profiling of MPNSTs and benign NF1 neurofibroma tissues - qPCR of differentially expressed in tumor tissues and MPNST cell lines - Lentiviral system for miR-204 trasnfection in NF1 and non-NF1 MPNST cell lines - Non-NF1 MPNST cells Xenograft | Gong et al. 2012 [38] |
| MSH2, MSH6, MSH3, MLH1, PMS2 | Phenotype overlapping between NF1 and Constitutional mismatch repair deficiency (CMMRD) Association with rare childhood malignancies | - Literature review about co-occurrence of symptoms and variants in genes associated with CMMRD and NF1 | Wimmer, Rosenbaum and Messiaen 2017 [39] |
| SDHB | Cause gastrointestinal stromal tumor (GISTs) | - SDHB expression by immunohistochemically in NF1-associated GISTs | Wang, Lasota and Miettinen 2011 [40] |
| Serotonin receptor 5′UTR 5-HT6 - HTR6 protein | Disrupting HTR6-neurofibromin interaction prevents agonist-independent HTR6-operated cAMP signaling in the prefrontal cortex, an effect that might underlie neuronal abnormalities in NF1 patients; 5-HT6 receptor may be considered as a potentially therapeutic target to correct some NF1-related cognitive deficits | - Nf1+/− heterozygote mice - HEK-293T and NG108-15 cell lines - Immunoprecipitation followed by Western blot analysis | Deraredj Nadim et al. 2016 [41] |
| SPRED1 nonsense, frameshift and missense mutations | Complete SPRED1 inactivation is needed to generate CALMs | - GWAS in unaffected and affected individuals. - SPRED1 cDNA sequencing - Melanocyte cell culture from normal skin and CALM of NF1 patient -Mouse embryonic fibroblasts | Brems et al. 2007 [42] |
| TERT mRNA and telomerase activity | Telomere dysfunction may play a role in driving genomic instability and clonal progression in NF1-associated MPNST | - High-resolution Single Telomere Length Analysis (STELA) of cutaneous and diffused plexiforme neurofibromas, and MPNSTs | Jones et al. 2017 [43] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woycinck Kowalski, T.; Brussa Reis, L.; Finger Andreis, T.; Ashton-Prolla, P.; Rosset, C. Systems Biology Approaches Reveal Potential Phenotype-Modifier Genes in Neurofibromatosis Type 1. Cancers 2020, 12, 2416. https://doi.org/10.3390/cancers12092416
Woycinck Kowalski T, Brussa Reis L, Finger Andreis T, Ashton-Prolla P, Rosset C. Systems Biology Approaches Reveal Potential Phenotype-Modifier Genes in Neurofibromatosis Type 1. Cancers. 2020; 12(9):2416. https://doi.org/10.3390/cancers12092416
Chicago/Turabian StyleWoycinck Kowalski, Thayne, Larissa Brussa Reis, Tiago Finger Andreis, Patricia Ashton-Prolla, and Clévia Rosset. 2020. "Systems Biology Approaches Reveal Potential Phenotype-Modifier Genes in Neurofibromatosis Type 1" Cancers 12, no. 9: 2416. https://doi.org/10.3390/cancers12092416
APA StyleWoycinck Kowalski, T., Brussa Reis, L., Finger Andreis, T., Ashton-Prolla, P., & Rosset, C. (2020). Systems Biology Approaches Reveal Potential Phenotype-Modifier Genes in Neurofibromatosis Type 1. Cancers, 12(9), 2416. https://doi.org/10.3390/cancers12092416