Simple Summary
Although the principle of personalized medicine has been the focus of attention, many cancer therapies are still based on a one-size-fits-all approach. The same holds true for targeting cancer cell survival mechanism that allows cancer cells to recycle their constituents (autophagy). In the past several indicators of elevated dependence of cancer cells on autophagy have been described. Addition of autophagy-inhibiting agents could be beneficial in treatment of these tumors. The biomarkers and mechanisms that lead to elevated dependence on autophagy are reviewed in the current manuscript.
Abstract
Recent advances in cancer treatment modalities reveal the limitations of the prevalent “one-size-fits-all” therapies and emphasize the necessity to develop personalized approaches. In this perspective, identification of predictive biomarkers and intrinsic vulnerabilities are an important advancement for further therapeutic strategies. Autophagy is an important lysosomal degradation and recycling pathway that provides energy and macromolecular precursors to maintain cellular homeostasis. Although all cells require autophagy, several genetic and/or cellular changes elevate the dependence of cancer cells on autophagy for their survival and indicates that autophagy inhibition in these tumors could provide a favorable addition to current therapies. In this context, we review the current literature on tumor (sub)types with elevated dependence on autophagy for their survival and highlight an exploitable vulnerability. We provide an inventory of microenvironmental factors, genetic alterations and therapies that may be exploited with autophagy-targeted approaches to improve efficacy of conventional anti-tumor therapies.
1. Introduction
Despite improvements in cancer therapy, cancers frequently relapse, illustrating the limitations of the current “one-size-fits-all” therapies. In part, this is related to the limited characterization of each individual tumor with its own genetic and phenotypic characteristics. Comprehensive analysis and identification of the tumor’s vulnerability will help to move forward into designed therapies with targeted treatments that are more effective in individual patients.
Genetic and microenvironmental inter- and intra-tumor differences in cancers result in biological reprogramming that increase reliance on specific signaling pathways for progression and survival. Identification of biomarkers that indicate pathway-addiction will aid in selecting the most effective treatment regimen between tumors of the same origin. This includes targeting of the “self-eating” mechanism, macroautophagy (hereafter, autophagy), which is frequently induced during cellular stress including hypoxia, nutrient deprivation and chemotherapy [1].
Autophagy is a homeostatic cellular catabolic process in which damaged and old cytoplasmic components (e.g., proteins and organelles) are sequestered into a double-membrane vesicle to be further degraded through lysosomal fusion. The products of lysosomal breakdown (e.g., amino acids and fatty acids) are recycled and transported across the lysosomal membrane into the cytoplasm. These metabolites can be reused in protein and ATP production [2]. During metabolic stress or periods of insufficient nutrient supply, autophagy can be rapidly upregulated to support energy production and provide building blocks for essential cellular functions.
For example, directly after birth, neonatal mice are exposed to nutrient starvation as the trans-placental nutrient supply has ceased. Autophagy enables survival to neonatal starvation through prevention of energy depletion until it is restored by milk intake [3]. In line, cells deprived of glucose or amino acids, or exposed to hypoxia, depend on autophagy for their survival [4,5,6]. In short, autophagy acts as a pro-survival mechanism through maintaining energy homeostasis and removal of toxic protein aggregates, pathogens and old/damaged organelles [7]. During conditions of starvation or metabolic stress, autophagy plays an important role by “recycling” amino acids and nutrients to maintain energy levels, protein synthesis and essential metabolic processes [8]. Furthermore, autophagy is essential for mitochondrial removal that might otherwise become cytotoxic [8,9,10,11,12].
In the context of cancer, (contextual) synthetic lethality is classically defined as the setting in which (in)activation of individual genes, nutrient status or microenvironment has limited effect on cell viability. The dependence on the supportive or compensating gene or mechanism results an attractive target in cancer therapy, mostly via pharmacological inhibition [13,14]. In contrast to normal cells, some cancer cells display elevated basal autophagy activity [15,16] and depend on autophagy for their survival [17,18]. Autophagy is highly responsive to nutrient availability and energy status [17,18], suggesting that tumors highly dependent on specific nutrients or have insufficient supply will be more responsive to autophagy-targeting therapies [19]. In the past years, several cellular alterations have been identified that increase the dependence of specific cancer cells on autophagy and may be important as biomarker in predicting the efficacy of autophagy inhibition in cancer therapy.
Combined targeting of autophagy with other targeted drugs could open a new window for further therapeutic strategies. However, the evidence that autophagy inhibition is an effective complementary strategy in cancer therapy is limited [20,21,22,23]. This may be related to lack of use of predictive biomarkers that identify autophagy-dependent tumors in patient populations and stresses the use of these biomarkers for future clinical trials.
In the past, selection of patient groups was frequently based on identifying increased autophagy activity, but this is not necessarily associated with elevated dependence on autophagy for survival. In addition, the role of autophagy (pro- or anti-survival) in cancer may be context dependent [24,25], highlighting the necessity to determine which tumor types and genetic/environmental/therapy backgrounds can synergize with autophagy targeting and are useful for patient stratification [26].
Here, we review cancer genotypes and metabolic statuses that increase cellular dependence on autophagy for survival. These include cancer cells with metabolic alterations (hypoxia and glutamine addiction), drug exposure (tyrosine kinase inhibitors), alterations due to changes in oncogenic signaling (e.g., MAPK, UPR and mTOR) or mutations (e.g., EGFRvIII and RAS). In addition, we list several emerging biomarkers that are associated with improved response to autophagy-inhibitory strategies (Table A1). These mechanisms and potential biomarkers illustrate a context dependent role for autophagy and advocate the principles of personalized medicine in trial design that include autophagy targeting.
2. Head and Neck Cancer
2.1. Hypoxia
The majority of solid human tumors, including glioblastoma, contain regions that are poorly oxygenated (hypoxia), a very heterogeneous and dynamic feature. Hypoxia is associated with a more malignant phenotype due to higher resistance to chemo- and radiotherapy and increased metastasis development. From a clinical point of view, reducing tumor hypoxia is highly desired since low oxygenation of tumors is associated with poor outcome in multiple cancer types [27]. Reduced oxygenation induces autophagy [6,28]. Although tumor hypoxia is a common feature of solid tumors and contributes to therapy resistance and disease progression in many cancer types, the evidence is strongest in head and neck cancer (HNSCC) [29]. In HNSCC, a meta-analysis indicated that the degree of hypoxia is the most significant factor explaining variability in survival [30] and demonstrated therapeutic benefit of hypoxia modification [31]. In multiple cancers, including HNSCC, expression of the autophagy related proteins microtubule associated protein 1 light chain 3 beta (LC3B), autophagy-related 5 (ATG5), unc-51-like autophagy activating kinase 1 (ULK1), BNIP and BCL2 interacting protein-like 3 (BNIP3L) is induced during hypoxia, indicating the importance for autophagy activation for survival [6,32,33]. Failure to induce these genes leads to autophagy inhibition and hypoxic cell death. In line, pharmacological targeting of autophagy results in elevated exposure to reactive oxygen species (ROS), a reduction in tumor hypoxia and sensitization of tumors to therapy, indicating that HNSCC tumors with high hypoxia will respond to autophagy inhibition [6,10,34]. Hypoxia associated patient stratification should therefore be considered before combining conventional therapies with autophagy-targeting.
2.2. PTOV1 Expression
Independent of hypoxia, recent studies have suggested that prostate tumor-overexpressed gene 1 (PTOV1) could serve as prognostic biomarker in nasopharyngeal and laryngeal tumors [35,36]. Expression of PTOV1 induces autophagy and is associated with increased resistance to cisplatin in vitro and in laryngeal cancer patients [37]. Targeting autophagy (hydroxychloroquine) in patient-derived cell lines that were cisplatin resistant (high PTOV1) resulted in vast cell killing, indicating the autophagy dependence and suggests that PTOV1 could be used as biomarker for selection of laryngeal cancer patients that would likely benefit from concurrent autophagy targeting.
3. Brain Cancer
3.1. EGFRvIII Expression
Glioblastoma (GBM) is the most malignant form of glioma and autophagy activation results in resistance to temozolomide [23,38], suggesting that autophagy targeting in combination with conventional therapy may have beneficial effects on therapy. A significant population of glioblastoma expresses a truncated EGFR receptor, epidermal growth factor receptor variant III (EGFRvIII), which is associated with increased hypoxia tolerance, radio- and chemoresistance and pro-survival [39]. Recently, we showed that ectopic and endogenous expression of EGFRvIII, in support of the high metabolic demand, increases dependence on autophagy for survival during hypoxia [34]. Adding the autophagy inhibitor chloroquine (CQ) reduces the viable hypoxic fraction and, subsequently, radiosensitized EGFRvIII GBM tumors. A previous study by Sotelo et al. indicated in a placebo controlled trial the clinical benefit of adding CQ to the treatment of surgery, carmustine and radiotherapy, extending patient survival to 24 months compared to 11 months for the control arm [40]. Retrospective analyses including patients of the same cohort indicated that patients with EGFRvIII+ tumors displayed the largest benefit. The incidence of EGFRvIII+ tumors is highly abundant (53%) in this cohort and may have contributed to the positive effect in the overall cohort [34]. Although not designed to test efficacy, patient stratification in a recently conducted phase 1 trial showed a similar trend that treatment of EGFRvIII+ glioblastoma benefit from combined treatment with CQ, temozolomide and radiotherapy [41].
A clinical trial conducted by Rosenfeld et al. did not yield a survival advantage in glioblastoma patients after targeting autophagy with hydroxychloroquine (HCQ) in combination with radiotherapy and temozolomide. Dose-limiting toxicity prevented escalation to higher doses of HCQ, and autophagy inhibition was not consistently achieved in patients treated with this regimen. In addition, the representation of EGFRvIII+ tumors has not been assessed [42]. Additionally, most drugs (including CQ and HCQ) are lysosomotropic drugs that accumulate in lysosomes due to the low PH. In general, the tumor microenvironment is acidic and may protonate autophagy inhibitors and prevent their diffusion over cellular membranes and limit their effectiveness [43].
One of the key regulators of autophagy activity is mammalian target of rapamycin (mTOR). Activation of mTOR supports cell proliferation and inhibits autophagy [44,45]. EGFR signaling stimulates activation of mTOR, resulting in an inverse relation of EGFR-expression to autophagy activity in cells. The residual autophagy is essential for cellular survival as the level of EGFR expression is associated with elevated cell killing after autophagy inhibition.
3.2. BRAFv600E
In addition to mutations and amplification of EGFR, abnormal activation of RAS/RAF/MEK/ERK pathway frequently occurs in cancer due to mutations in other membrane receptors, downstream effectors such as KRAS and BRAF or alternative pathways that regulate RAF activity such as phosphoinositide 3-kinase (PI3K), phosphatase and tensin homolog (PTEN) and protein kinase B (AKT) [46]. In high-grade pediatric central nervous system (CNS) tumors, BRAFV600E mutations are frequently observed (10–30% incidence) [47]. The expression of BRAFV600E drives autophagy activation and autophagy dependence for survival [48]. Combination of a BRAFV600E specific inhibitor, vemurafenib, with CQ synergistically improved outcome for a young patient diagnosed with a second recurrence of ganglioglioma [48]. The BRAFV600E mutation could be used as a selective marker to implement autophagy inhibitors in the treatment of childhood brain tumors [49]. In addition, despite an initial response, BRAF-driven cancer cells can become resistant to BRAFV600E kinase inhibition. Using early or late autophagy inhibitors (i.e., SBI-0206965, VPS34 knockdown, CQ, ATG5 knockdown and ATG7 knockdown) could overcome acquired resistance to BRAF inhibitors in brain tumor patients [50,51], indicating an important role for autophagy inhibition to prevent drug resistance.
3.3. Neurotrophins
Deregulation of growth factor receptors and aberrant secretion of their ligands contributes to malignancy of glioma [52]. Neurotrophins (NTs) and tropomyosin-related kinase receptor (Trk) are important in the development of the nervous system [53], but also contribute to tumor progression [54]. Jawhari et al. observed high expression of TrkC and NT-3 in sections of glioblastoma [55]. During periods of hypoxia, p38MAPK-induced TrkC/NT-3 signaling and autophagy are required to maintain mitochondrial homeostasis and promote glioblastoma cell survival. Expression of LC3B is most pronounced in hypoxic areas of the tumor [6]. In glioblastoma, high TrkC and NT-3 were associated with hypoxia and colocalized with LC3B and BNIP3. Targeting autophagy increased TrkC/NT-3 signaling and reduced proliferation, suggesting interdependence of these pathways. Combining CQ with TrkC-targeting resulted in vast cell killing, particularly in hypoxic cells [55]. Targeting autophagy in glioblastoma with elevated TrkC/NT-3 expression and/or signaling could be considered in future clinical trials.
3.4. WNT-Signaling
Many studies have suggested that WNT signaling is aberrantly activated in glioblastoma and that it promotes to glioblastoma growth and invasion via the maintenance of stem cell properties (reviewed in [56]). A recent study suggests that GBM harboring low WNT-CTNNB1 signaling or CTNNB1 mutations could benefit of autophagy inhibition [57]. CTNNB1/β-catenin is the principal effector of WNT canonical pathway and controls the expression of WNT target genes together with the transcription factor of the TCF/LEF families. In glioblastoma, hCTNNB1 mutations are rare and WNT signaling activation occurs mainly through upregulation of WNT ligands or epigenetic regulation of WNT inhibitors. Glioblastoma cell proliferation is maintained through canonical and non-canonical WNT signaling by the WNT pathway component disheveled segment polarity protein 2 (DLV2). Autophagy inhibits WNT signaling by promoting DLV2 and CTNNB1 degradation. In contrast, the TCF–CTNNB1 complex represses autophagy by increasing AKT/MAPK/mTOR signaling and by inactivating the transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and sequestosome 1 (SQSTM1) expression. Inhibition of WNT signaling through targeting TCF4 and autophagy flux reduces tumor growth, through a SQSTM1-dependent mechanism and caspase 8 activation. Combined targeting of WNT signaling and autophagy may therefore be beneficial in glioblastoma with high WNT signaling, but may also suggest that tumors with low WNT-CTNNB1 with high autophagy-SQSTM1 expression will be sensitive to autophagy inhibition [57].
In summary, high EGFR, EGFRvIII+ and BRAFV600E mutated glioblastoma and/or childhood tumors present a dependency on autophagy signaling. Autophagy targeting could be considered to improve outcome after therapy. In addition, TrkC and WNT-CTNNB1 expression status might identify potential glioblastoma susceptible to autophagy inhibitors in combination with targeting agents.
4. Pancreatic Cancer
Autophagy inhibition (HCQ) combined with gemcitabine/nab-paclitaxel is relatively well tolerated by patients, as demonstrated in preoperative phase 1–2 trials. Combination treatment improves pathological response, serum CA 19-9 levels and lymph node involvement and increases immune cell infiltration. However, the combined treatment does not improve overall and progression-free survival, indicating that autophagy could be an important therapeutic target in patients with pancreatic ductal adenocarcinoma (PDAC) but requires further optimization for optimal use in clinical contexts (NCT01128296 and NCT01978184 [58,59,60,61]). Several molecular markers and mechanisms in PDAC that have been linked to autophagy-addiction may be considered.
4.1. KRAS
PDAC are characterized with a high rate of KRAS mutations (approximately 95%), reprogrammed metabolism, hypervascular and hypoxic microenvironment and an invasive nature. Autophagy plays a critical role in KRAS-driven PDAC development and progression in response to hypoxia, nutrient deprivation and chemotherapy [62,63]. PDAC depend on autophagy to promote tumor proliferation and suppress cell death [16,64], support their nutritional requirements [65,66] and preserve metabolic homeostasis [15].
In late stage disease, KRAS mutation is often accompanied by a p53 loss of heterozygosity and high autophagy activity that dictates the progression of the pancreatic cancer. Mice developing KrasG12D/+, p53+/−, Atg5−/− PDAC display better overall survival. Blocking autophagy significantly compromises cell survival and patient-derived xenograft (PDX) growth independently of p53 mutational load [67]. Interestingly, autophagy impairment in the early stage of development (when p53 is still intact) may prevent the transformation of neoplastic pancreatic cancer towards invasive PDAC [68,69]. However, p53 has a dual role in regulating autophagy based on its cellular localization. In the nucleus, p53 activates autophagy through mTOR inhibition and AMP-activated protein kinase (AMPK) pathway, while cytoplasmic p53 inhibits autophagy, indicating that p53 status and localization has to be considered before the addition of autophagy inhibitors [70,71].
In addition, phospholipase D1 (PLD1) mediates autophagy [72] and modulates AMPK and mTOR (in) activation [73]. Suppression of AMPK results in increased PLD1 activity, resulting in increased phosphatidic acid (PA) production. Conversely, PLD1 inhibition increases ULK1 phosphorylation [73], and PLD1 knockdown results in inhibition of starvation induced autophagy [72], suggesting that PLD1 status may be used as biomarker to identify tumors that would benefit of targeting autophagy.
Nonetheless, other reports indicate that autophagy is dispensable to promote KRAS-driven cell growth [74,75], and autophagy addiction seems to be context-dependent. Cell type specific effects dictate whether oncogenic HRAS or KRAS mutations will stimulate or inhibit CQ-mediated toxicity [76]. This illustrates that oncogenic RAS determination is insufficient as a single biomarker to predict autophagy-inhibition efficacy and should only be considered in combination with additional biomarkers.
4.2. EI24 Expression
EI24 (etoposide-inducible autophagy associated transmembrane) is a p53-inducible gene that is frequently lost in breast and cervical cancers (17–41%). EI24 suppresses growth and functions as an effector of p53-mediated tumor progression. EI24 is an essential autophagy gene for execution of basal autophagy and clearance of aggregate-prone proteins [77]. Although the initial loss of EI24 and the associated loss of autophagy activity may have contributed to tumorigenesis [78], loss of EI24 at later stages impairs proliferation and cell survival in pancreatic cancer cells. In vivo, autophagy reduction through EI24 deficiency is not sufficient to significantly decrease tumor growth [79]. Although it is tempting to speculate that EI24-deficient tumors are unlikely to respond to autophagy inhibition, on several occasions we observed that autophagy targeting did not influence tumor growth, but was capable of inducing alterations in the tumor microenvironment that influence responsiveness to therapy (e.g., hypoxia) [6,34]. In addition, comparable to EGFR overexpressing glioblastoma, low autophagy activity, as observed in EI24 low cells, could increase the dependence of PDAC on residual autophagy. The potential of EI24 expression as biomarker for targeting autophagy in PDAC requires further investigation.
4.3. MAPK/ERK Inhibitors
Clinical use of MAPK/ERK inhibitors has shown some beneficial effects in the treatment of PDAC. Unfortunately, innate or acquired resistance is frequently observed and is a major limitation in effective use. In parallel, single autophagy inhibition in patients indicates limited efficacy and fails to improve outcome (NCT04132505 [80,81]). Noteworthy, in vitro data show promising response with the combination MAPK/ERK and autophagy inhibitors [82]. In KRAS WT or mutant tumors, pharmacological inhibition of ERK (SCH772984) or KRAS (ARS-1620) induces autophagy directly via increasing autophagy related gene expression and indirectly by altering cell metabolism (glycolysis) [83]. Autophagy inhibition synergizes with ERKi and enhances ERKi-mediated growth suppression in KRAS-mutant PDAC.
Notably, synergistic effects of KRAS/RAF/MEK/ERK inhibitors combined with autophagy inhibitors is not restricted to KRAS-driven PDAC models, but is also observed in NRAS-mutated melanoma, BRAF-mutated colorectal patient derived xenografts (PDXs) models [84] and BRAF-mutant thyroid cancers [85]. Kinsey et al. showed that patients benefit from the combination of MEK-inhibition (trametinib) with CQ after relapsing disease after first line treatment [84]. Additional clinical trials are currently determining the recommended dose of HCQ in combination with trametinib for future phase 2 trials in RAS mutant PDAC (NCT03825289 [86]).
4.4. Glutamine Addicted Cancer
Pancreatic cancer cells rewire intermediary metabolism to support different energetic and biosynthetic demands [87]. Alterations in nutrient uptake and catabolism, such as aberrant consumption of the nonessential amino acid glutamine, drive anabolic growth under hypoxic conditions and after oncogenic pathway dysregulation [88,89,90]. In absence of glucose, glutamine is the second main source of carbon to fuel the tricarboxylic acid cycle (TCA) cycle and the primary source of nitrogen for nucleotide and amino acid synthesis [79]. Through glutaminolysis, glutamine provides availability of nicotinamide adenine dinucleotide phosphate (NADPH), required for lipid synthesis and maintenance of the cellular redox balance. Insufficient delivery of glucose therefore increases the dependence on glutamine metabolism under lack of oxygen [91]. Glutamine limitation activates autophagy and in KRAS-driven PDAC, autophagy compensates the lack of glutamine via the degradation of intracellular macromolecules [92]. Conversely, blockage of autophagy lowers intracellular glutamine levels leading to cell apoptosis [19]. In line, we observed that HNSCC cancer cells that receive insufficient oxygen and glucose rapidly elevated autophagy activity. This provides the cell with liberation glutamine, as illustrated by the increased availability of non-protein associated extra- and intracellular glutamine during hypoxia (data not shown). The inability to active autophagy rapidly depletes intracellular glutamine [19], resulting in increased tumor cell killing and improved patient prognosis in several cancer types. Amino acids/glutamine transporters, solute carrier family 3 member 2 (SLC3A2/CD98hc) and solute carrier family 7 member 5 (SLC7A5/LAT1), expression could serve as markers of autophagy dependence. After chemo- and radiotherapy, absence of SLC3A2 impaired amino acid exchange and activation of mTOR/PI3K pathway to allow autophagy related gene expression. SLC3A2-deficient tumor cells rely on autophagy during glutamine deficiency and are radiosensitized after autophagy inhibition [93]. However, autophagy impairment during glutamine deprivation can reprogram the metabolic and transcriptional cell landscape, notably regulators of the TCA cycle [94] and may lead to increased expression of amino acids transporters (AATs) to compensate lack of glutamine and generate energy [18,95]. Blocking autophagy in combination with glutaminolysis inhibitor (e.g., C.968) has been shown to be an effective strategy in glutamine-addicted tumors [96].
In conclusion, inhibiting autophagy could synergize with current standard of care in KRAS mutant PDAC at early stage of development (p53 present), at late stage with p53 loss of heterozygosity and high autophagy activity or PDAC with low EI24 activity to enhance injurious effect. This therapy could be extendable to autophagy addicted RAS-driven cancers [24] and glutamine-addicted (SLC3A2-deficient) PDAC and HNSCC tumors.
5. Melanoma
BRAFv600E
The BRAFV600E mutation is the most common BRAF alteration (80%) and an important oncogenic driver in melanomas, pediatric CNS, lung cancer (3%) and colorectal cancer (6–12%) [97,98]. Melanomas present the highest alteration rate with over 90% of BRAFV600E mutations, accompanied with 30–40% mutations in PTEN. Autophagy is essential for survival of cells with BRAFV600E mutations [48,99,100], maintenance of lung cancer cell metabolic homeostasis [101,102] and development of treatment resistance in colorectal cancer cells [103].
In genetically engineered mouse models (GEMMs) that yield BrafV600E- and Pten-deficient melanomas, tumor-specific ablation of Atg7 resulted in accumulation of defective mitochondria and impaired tumor growth. Pharmacological targeting of BRAFV600E (dabrafenib) enhances the anti-proliferative effect [104], indicating that autophagy inhibition in combination with BRAFV600E inhibitors could provide an effective strategy in treatment of melanomas. The treatment of melanomas BRAFV600E inhibitor has proven to be an effective therapy. Nevertheless, melanoma frequently relapses due to acquired resistance. Ma et al. identified that melanomas with highest resistance to BRAFV600E inhibitors displayed elevated levels of autophagy. In vitro inhibition of BRAFV600E resulted in activation of the unfolded proteins response due to sequestration of BRAFV600E in the endoplasmic reticulum and resulted in activation of cytoprotective autophagy. Inhibition of autophagy in combination with BRAFV600E targeting resulted in tumor regression [105]. Considering the encouraging results, phase 1 and 2 clinical trials have been initiated that combine BRAFV600E inhibitors or MEK inhibitors (downstream of BRAF-signaling) with HCQ (NCT01897116 [106] and NCT02257424 [107]).
6. Colorectal Cancer
6.1. KRAS
KRAS mutations account for approximately 30–50% of oncogenic driving mutations in colorectal cancer (CRC). Similar to BRAFV600E, KRAS alterations induce autophagy through MEK/ERK signaling and contribute to CRC cell survival [108]. Comparable to observations in pancreatic cancer, genetic ablation of autophagy in KRAS-driven CRC tumors can either promote or inhibit tumor growth driven by activation of different signaling pathways. Regressing autophagy-deficient tumors display an induction of the NOTCH signaling and a reduction in mitochondrial activity. Tumors that grow despite autophagy ablation rely on PI3K/AKT/mTOR signaling and a decrease in glucose metabolism for their expansion [109].
Signal transduction of KRAS-mutant CRC cell line is dependent on effective RAF networks. Lee et al. identified BRAF, CRAF and ATG7 as the best target combination to minimize toxicity in WT KRAS cells and normal tissue effects but maximize the effect in the KRAS-mutated cells. Co-targeting autophagy and MAPK pathways, by silencing ATG7 together with therapeutic agents against MEK, BRAF and CRAF (trametinib and RAF709), spared normal cells, while inducing apoptosis in KRAS mutated CRC and also PDAC cells [110].
6.2. JNK-Signaling
Similar to other MAPKs, the c-Jun N-terminal kinase (JNK) proteins are activated by a series of phosphorylation events in response to multiple stimuli such as cytokines, growth factors, pathogens, stress, toxins and drugs to promote tumorigenesis. During hypoxia, the PKCδ (protein kinase Cδ)/JNK1 axis promotes autophagy through Bcl-2 phosphorylation followed by dissociation from Beclin1 [111]. JNK1 inhibition impaired hypoxia-induced autophagy and resulted in increased cell death [112]. The validity of JNK1 as potential biomarker for autophagy-dependence is currently being evaluated in a phase 1/2 clinical trial in CRC (NCT01206530 [113]).
6.3. TFEB
Lysosomal interaction is an essential part of autophagy. TFEB regulates transcription of lysosomal genes and can be trailed through its translocation from the cytoplasm to the nucleus [114]. In non-small cell lung cancer (NSCLC), TFEB presence is associated with high levels of lysosomal markers and a poor prognosis [115]. In CRC, nuclear TFEB drives doxorubicin-induced autophagy and prevents drug-associated apoptosis. Genetic or pharmacological inhibition of autophagy partially reverses drug-resistance and results in increased cell death [116].
In short, driver mutations in BRAFV 600E or KRAS, JNK1 expression and nuclear localization of TFEB are indicative of CRC tumors that are susceptible to autophagy targeting and clinical evaluation should be considered.
7. Breast Cancer
7.1. JAK/STAT Signaling
Autophagy is important in mammary tumorigenesis [117,118] and alterations in important autophagy-regulatory effectors such as PI3K, p53, EGFR and Bcl-2 proteins; PI3K/mTOR signaling [119]; and Janus kinases–signal transducer and activator of transcription proteins (JAK/STAT) pathways are frequently observed [120]. Constitutive activation of STAT3 signaling is observed in 50–60% of breast cancer events. This pathway is key in transducing extracellular signals (cytokines, interferons or growth factors) into transcriptional programs that regulate cell growth, invasiveness, metastasis and maintenance of cancer stem cell (CSC) viability [120,121]. Autophagy inhibition particularly affects stem cells, cytokine secretion and epithelial-to-mesenchymal transition (EMT) in breast cancer cells. Lacking a leader sequence [122], autophagy controls IL6 secretion and is required for CSC viability [123]. Interestingly, elevated production of interleukin 6 (IL6) correlates with increased p-STAT3 and contributes to the paracrine activation of signal transducer and activator of transcription 3 (STAT3) [124]. Breast cancer cells highly active in STAT3 activity display increased dependency on autophagy for their survival. Advantageously, combining autophagy inhibitors with doxorubicin synergistically inhibits tumor growth in triple-negative breast cancer (TNBC) with high JAK/STAT pathway activation, advocating for autophagy inhibition in this subtype of tumors [125].
7.2. HER2 Signaling
HER2 (human epidermal growth factor receptor 2) is a transmembrane receptor tyrosine kinase (RTK) of the EGFR family and HER2 inhibition is a common strategy for breast cancers with HER2 amplification. Transphosphorylation of the protein dimer stimulates downstream signaling, including MAPK cascades, PI3K/AKT/mTOR pathway or STAT transcription factors, all known regulators of autophagy [126]. Administration of RTK inhibitors induces autophagy, which supports the development of resistance mechanisms [127,128]. After acquisition therapy resistance, HER2+ tumors maintain a higher basal level of autophagy to support their proliferation [129,130]. Recent preclinical studies indicate that autophagy inhibition with HER2-targeted inhibitors may enhance tumor cell death in breast cancer cells as autophagy impairment restores sensitivity to HER2 inhibitors in breast cancer and in lapatinib-resistant EAC cell line (HER2-amplified esophageal adenocarcinoma) [131,132,133]. In line, monoallelic loss of BECN1 is frequent in HER2+ breast cancers and predicts response to HER2-inhibitors (trastuzumab) [134]. BECN1 overexpression enhances HER2 phosphorylation/activation and decreases response to RTK inhibitors (lapatinib). Conversely, Beclin1 knockdown improved lapatinib-induced apoptosis [135], indicating that HER2 treatment efficacy is largely determined by the autophagy-status of the cell. Targeting HER2 signaling in combination with autophagy inhibitors is an appealing strategy.
Phase 1 and 2 clinical trials are currently ongoing to evaluate the asset of autophagy inhibitors alone [136,137,138] or together with other chemotherapeutic agents, such as microtubules inhibitors (taxane and taxane-like drugs) [139], mTOR inhibitor (everolimus) [140] and PI3K/mTOR inhibitors (gedatolisib) [141]) in breast cancer treatment.
7.3. Estrogen Receptor Signaling
Estrogen receptor-positive (ER+) breast cancer is effectively treated with endocrine therapies. In response to endocrine therapy, autophagy is induced and contributes to resistance to anti-hormone therapies [142,143,144]. In line, tamoxifen-resistant breast cancer cells display high autophagy activation combined with overexpression of metastasis-associated 1 (MTA1). In ER+ breast cancer patients treated with tamoxifen, MTA1 expression increases in relapsed or recurrent disease and correlates with low disease free survival. Knockdown of MTA1 decreases autophagic activity, indicating a direct role for MTA1 in regulation of autophagy. Importantly, pharmacological or genetic inhibition of autophagy restores tamoxifen sensitivity, indicating that autophagy inhibitory strategies could be considered for tamoxifen resistant, high MTA1 expressing breast cancer [145].
7.4. CDK4 and-6 Inhibition
Alternatively, ER+/HER2− breast cancers may be treated with cyclin-dependent kinase CDK4 and CDK6 inhibitors (e.g., palbociclib, abemaciclib or ribociclib). CDK4/CDK6/cyclin D signaling is altered in 35% of ER+ breast cancer patients, but 16% of patients fail to respond to the treatment. CDK4/CDK6 inhibition increases autophagy. Interestingly, the inhibition of CDK4/CDK6 in combination with genetic (siRNA Beclin1 or siRNA ATG5) or pharmacological (HCQ, spautin-1 or bafilomycin A1) autophagy targeting induces a synergistic anti-proliferative effect in vitro and diminishes growth of patient-derived xenografts isolated from retinoblastoma-associated protein positive (Rb+) and low-molecular-weight isoforms of cyclin E (LMWE) cytoplasmic negative (LMWE cyt−) breast cancer [146]. In summary, LMWE appears to be predictive in sensitivity for the combination of CDK4/CDK6 inhibitors with autophagy targeting in advanced ER+ breast cancer [146].
7.5. IKBKE and JMJD6
Both inhibitor of nuclear factor kappa B kinase subunit epsilon (IKBKE) and Jumonji domain-containing 6 (JMJD6) contribute to enhancement of tumor supportive autophagy. In a myristoylated screen, IKBKE was identified as an inducer of autophagy in triple negative and in HER2-positive breast cancers. Active IKBKE-induced autophagy supports oncogenic cellular transformation of immortalized mammary cells and is required for TNBC cell proliferation, suggesting that targeting autophagy represents a feasible approach to prevent TNBC evolution and progression [147].
JMJD6 overexpression results in increased malignancy, such as increased migration and poor differentiation that eventually leads to poor prognosis. A newly discovered intrinsic tyrosine kinase domain in JMJD6 is required for H2AX Y39 phosphorylation and mediates transcriptional regulation of ATG genes in TNBC. Impairment of kinase activity and autophagy suppresses TNBC growth in vivo, implying that JMJD6 could serve as predictive biomarker for autophagy inhibition in breast cancer [148].
In summary, targeting autophagy appears to be a profitable approach in several breast cancer subtypes. In HER2+, autophagy inhibition reverses acquired tyrosine kinase inhibitors (TKIs) resistance and enhances cell mortality. IKBKE activation identifies HER2+ as well as TNBC tumors susceptible to autophagy inhibition. In TNBC, treatment opportunities remain limited, although could benefit from autophagy inhibition when displaying high STAT3 activity or high JMJD6 expression. Finally, MTA1 may identify receptive tamoxifen-ER+ resistant cells and their dependence to autophagy pathway.
8. Lung Cancer
8.1. TKI
In NSCLC, activating mutations in EGFR are favorable predictors for EGFR TKIs-based therapies. Although EGFR TKIs can prolong survival, different acquired resistance mechanisms have been identified. Although the degree of induction varies according to the drug and the cancer cell lines [149,150], autophagy activation is systematic and recurrent upon TKIs independently of the target and the cancer type. Cytoprotective autophagy has been observed in mutant and wild-type EGFR NSCLC [150,151,152] and in EGFR mutant bladder cancer [153]. Inhibiting autophagy-mediated TKI-resistance may be considered as therapeutic strategy to overcome resistance TKIs in EGFR deregulated cancers [21,82,154,155]. Considering the positive pre-clinical data, addition of autophagy inhibitor (HCQ) to standard of care erlotinib is currently being evaluated in patients with mutant or high expressing EGFR NSCLC tumors (NCT00977470 [156]).
8.2. KRAS and LKB1 Signaling
Similar to PDAC, autophagy plays an important role in KRAS- and p53-driven lung cancer tumorigenesis and metabolism [157,158]. In Western countries, KRAS and liver kinase B1 (LKB1 or STK11) are the most common oncogene driver mutations in NSCLC (~40% of cases) [159,160]. Tumor suppressor gene LKB1 encodes an upstream kinase of AMPK, which enables the dissociation between ULK1 and mTOR thereby allowing ULK1 association with ATG13/FIP200 to activate autophagy [44,161]. Loss of LKB1 reprograms lung cancer metabolism to generate energy and sustain cell proliferation. Upon starvation, LKB1 mutant human lung cancer cells depend on autophagy to maintain energy homeostasis by recycling substrates for TCA metabolism and biogenesis of fatty acids. Moreover, autophagy is required for tumor initiation and progression and energy homeostasis in Lkb1-deficient Kras-driven NSCLC GEMMs. Atg7 and Lkb1 loss sensitize tumor-derived cell lines (TDCLs) to metabolic stress, suggesting that autophagy inhibition can be a favorable approach for the treatment of Lkb1-deficient lung tumors [162,163].
In summary, acquired resistance to TKIs in EGFR wild-type and mutant tumors (e.g., HER2+ breast, bladder and lung) is (partially) reversed by autophagy inhibitors. Lkb1 deficiency identifies tumors that are highly vulnerable to autophagy-inhibition due to their sole dependence on autophagy to meet its metabolic demand.
9. Liver Cancer
HGF Signaling
Hepatocyte growth factor (HGF) and MET signaling supports cellular proliferation, promotes epithelial-mesenchymal transition (EMT) and causes invasion and metastasis during malignant transformation in hepatocellular carcinoma (HCC). MET kinase activity plays a key role in cancer cell metabolism through induction of glycolysis, glutamine synthesis through increased glutaminase activity, glutaminolysis and lipid synthesis [164]. Interestingly, MET contains two distinct LC3-interacting regions (LIRs) close to two tyrosine phosphorylation sites and orchestrates the autophagic response. Dephosphorylated/inactive MET interacts with LC3 and promotes autophagy activity. In 51% of HCC patients, dephosphorylated MET associated with absence of SQSTM1 was observed, suggesting SQSTM1 degradation through high autophagy activity. Combined targeting of MET-activation and autophagy synergistically reduces cell viability and results in HCC regression. These results suggest that TKI treatment of HCC with high HGF-MET signaling activity and autophagy-targeting should be considered for effective therapy [164].
Autophagy is essential for CD24-mediated resistance to sorafenib. CD24 is glycoprotein that is highly expressed in tumors including HCC and mediates kinase inhibitor resistance through PP2A/AKT/mTOR inactivation and activation of lysosomal degradation. Blocking autophagy with bafilomycin A1 or knocking-down ATG5 overcomes sorafenib resistance in CD24 high expressing HCC, suggesting that CD24 could be used as a biomarker for therapeutic benefit for autophagy-inhibition in sorafenib-resistant HCC [165].
10. Prostate Cancer
KDMB4 Expression
Pten and Atg7-autophagy loss in GEMMs revealed tumor-promoting effects of autophagy in castrate-naïve and castrate-resistant prostate cancers (PCa) [166]. The histone lysine demethylase KDM4B fulfills a critical role in this signaling by co-activating androgen and estrogen receptors [167,168]. In PCa, KDM4B stimulates androgen receptor (AR) transcription activity via histone demethylation and AR ubiquitination [169]. High KDM4B expression correlates with resistance to androgen deprivation treatment in PCa patients [170]. In castration-resistant prostate cancer (CRPC) cells, KDM4B expression stimulates autophagy activation through Wnt/beta-catenin signaling and sequestration of beta-catenin in the nucleus. Autophagy inhibition reverses the growth advantage of KDM4B expressing cells, suggesting that high KDM4B expression identifies CRPC patients that could benefit from autophagy targeted therapy [170].
11. Bone Cancer
HSP90AA1
Bone cancers are rare at less than 1% of all cancers. Osteosarcoma and chondrosarcoma are the two main subtypes of primary bone malignancies. Chemotherapy is generally not very effective for chondrosarcoma, while it is an important part of the treatment for osteosarcoma. Therapy resistance can arise through expression of conserved chaperone heat shock proteins (HSPs). HSP90AA1-induced doxorubicin-resistance inhibits cell death through deactivation of JNK and p38 kinase signals and stimulates pro-survival autophagy through PI3K/AKT/mTOR signaling inhibition. Hence, targeting HSP90AA1 or autophagy recovery restores sensitivity to chemotherapy induced killing. These results suggest that HSP90AA1high osteosarcoma could benefit from concurrent autophagy inhibition [171].
12. Conclusions
As observed with other new discoveries and therapeutic intervention possibilities in oncology (e.g., immunotherapy, apoptosis and extracellular vesicles), the “one size fits all” approach does not apply to autophagy inhibition as well. Although there are some common mechanisms or microenvironmental stresses that increase the dependence on autophagy for survival, intrinsic differences determine the efficacy of autophagy inhibitory strategies. Identification of biomarkers that predict autophagy-addiction in combination with autophagy inhibition appears to be the most logical and most effective strategy.
In this context, we review the current literature and list the different cancer (sub)types that pre-clinically or in more advanced stages have been associated with increased dependence on autophagy for survival. These include genetic changes, e.g., EGFR, EGFRvIII and BRAFv600E, that increase dependence on autophagy to supply in the high metabolic demand and chemotherapy-resistance mechanisms, e.g., against TKI and chemotherapy (Figure 1 and Figure 2). The assessment of autophagy dependence is however complex and is often assumed to reveal itself by increased autophagy activity. Nevertheless, an important point is that high autophagy flux does not guarantee an addiction to the signaling and can be seen as a “side effect” of other pathway alterations without contributing to cells survival. In addition, low autophagy activity in some cases dictates sensitivity to autophagy inhibitors [172], most likely due to high dependence on the residual autophagy activity to maintain cellular homeostasis. Unfortunately, in most cancer (sub)types where reduced autophagy activity is observed, no assessment of autophagy-dependence will be done.

Figure 1.
Genetic changes and chemotherapy-resistance mechanisms that contribute to elevate autophagy dependence. Biomarkers in grey require further investigation (created using Https://Biorender.com).
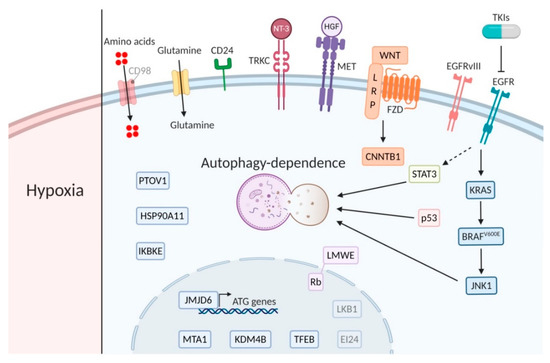
Figure 2.
Major players of the main signaling pathways. Bright biomarkers indicate amplification or active signaling and blurred biomarkers indicate low activity or expression in relation to autophagy dependence (created using Https://Biorender.com).
Clinical trials in different cancer types have been conducted and demonstrated the relative safety of targeting autophagy with CQ or HCQ. Nevertheless, several trials have not been able to provide patients with the maximum intended dose before reaching the maximum tolerated dose [42]. To date, it remains impossible to measure autophagy inhibition in patients at the site where autophagy inhibition is intended, the tumor, without taking sequential biopsies. In most circumstances, this is not possible or undesired due to the associated risk and extra burden for the patient. Whether sufficient autophagy inhibition in the tumor is reached to successfully increase therapy efficacy remains a matter of investigation. Without having tools available that indicate whether sufficient autophagy inhibition is reached, the evaluation of the success of autophagy-inhibitory strategies will be dependent on improving outcome for patients after therapy. Patient stratification and selection for tumor types with biomarkers that have shown promising results in preclinical studies will determine the feasibility of autophagy inhibition in oncological care.
Author Contributions
L.M.O.B. reviewed and analyzed available literature and wrote the manuscript, T.G.H.K. wrote the manuscript, K.M.A.R. designed the scope of the review and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
These studies received financial support from the Dutch Cancer Society (KWF grant 12,276 and UM2015-7735 to K.R.) and Zeldzame Ziekten Fonds (ZZF grant to K.R.).
Acknowledgments
The authors would like to apologize to the authors whose work could not be included due to the extensive field of research.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

Table A1.
Tumor (sub)types and biomarkers associated with autophagy-dependence and their improved combination therapies.
Table A1.
Tumor (sub)types and biomarkers associated with autophagy-dependence and their improved combination therapies.
| Cancer Type | Tumor Type | Biomarker/Factor | Mechanism of Autophagy Dependency (When Known) | References |
|---|---|---|---|---|
| Head and neck cancer | HNSCC | Hypoxia | Nutrient suppletion, reduction ROS exposure | [6,10,33] |
| Laryngeal cancer cisplatin or 5-FU resistant | PTOV1high | Cisplatin resistance | [37] | |
| HNSCC | EGFRhigh | - | ||
| HNSCC | Glutamine | Glutamine suppletion | NP* | |
| HNSCC | SLC3A2- | - | [93] | |
| Esophageal HER2 amplification | TKIs against EGFR and HER2 | - | [133] | |
| Brain cancer | Hypoxic glioblastoma | EGFRvIII+ | Nutrient suppletion | [34] |
| Glioblastoma | EGFRhigh | - | [172] | |
| Central nervous system | BRAFV600E | - | [47,48] | |
| Gliomas | TRKC/NT-3high | Hypoxia tolerance | [55] | |
| Glioblastoma | WNT-CTNNB1low or mutated | - | [57] | |
| Neuroblastoma | TKIs against EGFR or multiple TKIs or Axl | - | ||
| Pancreatic cancer | Kras-driven | Kras | - | [16,64] |
| - | [67] | |||
| - | [84] | |||
| PDAC | EI24low | - | [79] | |
| KRAS-driven | Glutamine | Glutamine suppletion | [19] | |
| Thyroid cancer | BRAFV600E-driven | BRAFV600E inhibitor | - | [85] |
| Skin cancer | Melanoma | BRAFV600E | Removal of mutated protein aggregates | [104] |
| Colorectal cancer | CRC | BRAFV600E | - | [103] |
| KRAS-driven | KRAS | - | [110] | |
| CRC, metastatic CRC or adenocarcinoma | JNK1+ | - | [113] | |
| Doxorubicin resistant | TFEB+ | - | [116] | |
| Breast cancer | TNBC | STAT3 | - | [125] |
| HER2+ | TKIs against HER2 | TKI resistance | [131] | |
| TKIs against EGFR and HER2 | TKI resistance | [132] | ||
| ER+ breast cancer tamoxifen resistant | MTA1high | Tamoxifen resistance | [145] | |
| Breast cancer with Rb+ and LMWE cyt- or Letrozole or Anastrozole resistant | Rb+ and LMWE cyt- | - | [146] | |
| TNBC and HER2- | IKBKEhigh | - | [147] | |
| TNBC | JMJD6high | JMJD6 phosphorylates H2A.XY39 and regulates ATG genes transcription | [148] | |
| Lung cancer | BrafV600E-driven | BrafV600E | - | [101] |
| NSCLC with EGFR WT and mutant | TKIs against EGFR | - | [151] | |
| - | [152] | |||
| Kras- and p53-driven | Kras | - | [157,158] | |
| Lkb1-deficient Kras-driven NSCLC | Lkb1 (STK11) | TCA and fatty acid synthesis support | [163] | |
| Bladder cancer | EGFR mutant | TKIs against EGFR and HER2 | - | [153] |
| Liver cancer | HCC | HGF-METhigh | - | [164] |
| HCC sorafenib resistant | CD24high | Sorafenib resistance | [165] | |
| Prostate cancer | Castration-resistant | KDM4Bhigh | - | [170] |
| Bone cancer | Osteosarcoma doxorubicin or cisplatin or methotrexane resistant | HSP90A11high | Doxorubicin resistance | [171] |
*NP: non-published observation.
References
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Marx, J. Autophagy: Is it cancer’s friend or foe? Science 2006, 312, 1160–1161. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Role of Autophagy in Cancer. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.; Beucken, T.V.D.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2009, 120, 127–141. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12, 1535–1541. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Boil. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Rouschop, K.; Ramaekers, C.H.; Schaaf, M.B.; Keulers, T.G.; Savelkouls, K.G.; Lambin, P.; Koritzinsky, M.; Wouters, B. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, X.; Yuan, F.; Zhu, W.-G.; Zhao, Y. Autophagy-deficient tumor cells rely on extracellular amino acids to survive upon glutamine deprivation. Autophagy 2018, 14, 1652–1653. [Google Scholar] [CrossRef]
- Seo, J.-W.; Choi, J.; Lee, S.-Y.; Sung, S.; Yoo, H.J.; Kang, M.-J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.-D.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; LaRocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2019, 27, 843–857. [Google Scholar] [CrossRef]
- Wouters, B. Targeting hypoxia tolerance in cancer. Drug Resist. Updat. 2004, 7, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.; Hill, R.; Tannock, I.F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar] [CrossRef]
- Overgaard, J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck-A systematic review and meta-analysis. Radiother. Oncol. 2011, 100, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Boil. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Cojocari, D.; Keulers, T.G.; Jutten, B.; Starmans, M.H.; De Jong, M.C.; Begg, A.C.; Savelkouls, K.G.; Bussink, J.; Vooijs, M.; et al. The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother. Oncol. 2013, 108, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.E.M.G.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Lin, H.; Wu, S.; Lei, F.; Zhu, X.; Song, L.; Hong, M.; Guo, L. Prostate Tumor Overexpressed 1 (PTOV1) Is a Novel Prognostic Marker for Nasopharyngeal Carcinoma Progression and Poor Survival Outcomes. PLoS ONE 2015, 10, e0136448. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Wang, Y.; He, Z.; Chen, H.; Liang, S.; He, S.; Wu, S.; Song, L.; Chen, Y. Prostate tumor overexpressed-1, in conjunction with human papillomavirus status, predicts outcome in early-stage human laryngeal squamous cell carcinoma. Oncotarget 2016, 7, 31878–31891. [Google Scholar] [CrossRef]
- Mayea, Y.G.; Mir, C.; Muñoz, L.; Benavente, S.; Castellví, J.; Temprana, J.; Maggio, V.; Lorente, J.; Paciucci, R.; E Lleonart, M. Autophagy inhibition as a promising therapeutic target for laryngeal cancer. Carcinogenesis 2019, 40, 1525–1534. [Google Scholar] [CrossRef]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef]
- Theys, J.; Jutten, B.; Dubois, L.; Rouschop, K.; Chiu, R.K.; Li, Y.; Paesmans, K.; Lambin, P.; Lammering, G.; Wouters, B. The deletion mutant EGFRvIII significantly contributes to stress resistance typical for the tumour microenvironment. Radiother. Oncol. 2009, 92, 399–404. [Google Scholar] [CrossRef]
- Sotelo, J.; Bricenño, E.; Loópez-Gonzaález, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2020, in press. [Google Scholar]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hägg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nature 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascades Which Alter Therapy Response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef]
- Dahiya, S.; Emnett, R.J.; Haydon, D.H.; Leonard, J.R.; Phillips, J.J.; Perry, A.; Gutmann, D.H. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. 2013, 16, 318–319. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity in BRAFV600E Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Foreman, N.K.; Thorburn, A. Using BRAF(V600E) as a marker of autophagy dependence in pediatric brain tumors. Autophagy 2014, 10, 2077–2078. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, 358. [Google Scholar] [CrossRef]
- Zahedi, S.; Fitzwalter, B.E.; Morin, A.; Grob, S.; Desmarais, M.; Mellan, A.; Green, A.L.; Vibhakar, R.; Hankinson, T.C.; Foreman, N.K.; et al. Effect of early-stage autophagy inhibition in BRAF(V600E) autophagy-dependent brain tumor cells. Cell Death Dis. 2019, 10, 679. [Google Scholar] [CrossRef] [PubMed]
- Hamel, W.; Westphal, M. Growth factors in gliomas revisited. Acta Neurochir. 2000, 142, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, J.C.; Wu, S.H. Neurotrophin signaling: Many exciting surprises! Cell. Mol. Life Sci. 2006, 63, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, G.M.; Choi, Y.-J.; Kim, H.J.; Kim, Y.-J.; Jin, W. TrkC Promotes Survival and Growth of Leukemia Cells Through Akt-mTOR-Dependent Up-Regulation of PLK-1 and Twist-1. Mol. Cells 2013, 36, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Jawhari, S.; Bessette, B.; Hombourger, S.; Durand, K.; Lacroix, A.; Labrousse, F.; Jauberteau, M.-O.; Ratinaud, M.H.; Verdier, M. Autophagy and TrkC/NT-3 signaling joined forces boost the hypoxic glioblastoma cell survival. Carcinogenesis 2017, 38, 592–603. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2015, 96, 137–150. [Google Scholar] [CrossRef]
- Nager, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Study of Pre-surgery Gemcitabine + Hydroxychloroquine (GcHc) in Stage IIb or III Adenocarcinoma of the Pancreas. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT01128296?term=NCT01128296&draw=2&rank=1 (accessed on 17 July 2020).
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; A Liotta, L.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Randomized Phase II Trial of Pre-Operative Gemcitabine and Nab Paclitacel With or With Out Hydroxychloroquine. 2020. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01978184?term=NCT01978184&draw=2&rank=1 (accessed on 17 July 2020).
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.-I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. Autophagy in pancreatic cancer pathogenesis and treatment. Am. J. Cancer Res. 2012, 2, 383–396. [Google Scholar] [PubMed]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Boil. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; RajeshKumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.; Herman, J.M. Autophagy, p53, and Pancreatic Cancer. N. Engl. J. Med. 2014, 370, 1352–1353. [Google Scholar] [CrossRef]
- Levine, B.; Abrams, J.M. p53: The Janus of autophagy? Nature 2008, 10, 637–639. [Google Scholar] [CrossRef]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Hurtado-Lorenzo, A.; Tian, H.; Morel, E.; Nezu, A.; Chan, R.B.; Yu, W.H.; Robinson, K.S.; Yeku, O.; Small, S.A.; et al. The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 2010, 1, 1–11. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frías, M.A.; Foster, D.A. Reciprocal Regulation of AMP-activated Protein Kinase and Phospholipase D*. J. Boil. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2015, 113, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Eng, C.H. Revisiting autophagy addiction of tumor cells. Autophagy 2016, 12, 1206–1207. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Gamez, G.; Menke, C.; Hernandez, A.; Thorburn, J.; Gidan, F.; Staskiewicz, L.; Morgan, S.; Cummings, C.; Maycotte, P.; et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014, 10, 1814–1826. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhao, H.; Miao, L.; Wang, L.; Sun, F.; Zhang, H. The p53-induced GeneEi24Is an Essential Component of the Basal Autophagy Pathway. J. Boil. Chem. 2012, 287, 42053–42063. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.-S.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; A Gewirtz, D.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Hwang, M.; Jun, D.W.; Kang, E.H.; Yoon, K.-A.; Cheong, H.; Kim, Y.-H.; Lee, C.-H.; Kim, S. EI24, as a Component of Autophagy, Is Involved in Pancreatic Cell Proliferation. Front. Oncol. 2019, 9, 652. [Google Scholar] [CrossRef]
- Binimetinib and Hydroxychloroquine in Treating Patients With KRAS Mutant Metastatic Pancreatic Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04132505?term=NCT04132505&draw=2&rank=1 (accessed on 17 July 2020).
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Jutten, B.; Rouschop, K. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2013, 13, 42–51. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 2016, 102, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Trametinib and Hydroxychloroquine in Treating Patients With Pancreatic Cancer (THREAD). 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03825289?term=NCT03825289&draw=2&rank=1 (accessed on 17 July 2020).
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Boil. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Digomann, D.; Kurth, I.; Tyutyunnykova, A.; Chen, O.; Löck, S.; Gorodetska, I.V.; Peitzsch, C.; Skvortsova, I.-I.; Negro, G.; Aschenbrenner, B.; et al. The CD98 Heavy Chain Is a Marker and Regulator of Head and Neck Squamous Cell Carcinoma Radiosensitivity. Clin. Cancer Res. 2019, 25, 3152–3163. [Google Scholar] [CrossRef]
- Lin, T.-C.; Chen, Y.-R.; Kensicki, E.; Li, A.Y.-J.; Kong, M.; Li, Y.; Mohney, R.P.; Shen, H.-M.; Stiles, B.; Mizushima, N.; et al. Autophagy: Resetting glutamine-dependent metabolism and oxygen consumption. Autophagy 2012, 8, 1477–1493. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, X.; Yuan, F.; Zhang, L.; Wang, Y.; Wang, L.; Mao, Z.; Luo, J.; Zhang, H.; Zhu, W.-G.; et al. Increased Amino Acid Uptake Supports Autophagy-Deficient Cell Survival upon Glutamine Deprivation. Cell Rep. 2018, 23, 3006–3020. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, L.-Q.; Huang, J.-F.; Liu, K.; Chuai, Z.-R.; Yang, Z.; Wang, Y.-X.; Shi, D.-C.; Liu, Q.; Huang, Q.; et al. BRAF Mutations in Patients with Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.; Morgan, M.J. Targeting Autophagy in BRAF-Mutant Tumors. Cancer Discov. 2015, 5, 353–354. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Targeting Autophagy Addiction in Cancer. Oncotarget 2011, 2, 1302–1306. [Google Scholar] [CrossRef]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.M.; Hu, J.; Zhang, G.; et al. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Trial of Vemurafenib and Hydroxychloroquine in Patients With Advanced BRAF Mutant Melanoma. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01897116?term=NCT01897116&draw=2&rank=1 (accessed on 17 July 2020).
- Dabrafenib, Trametinib and Hydroxychloroquine in Patients With Advanced BRAF Mutant Melanoma (BAMM). 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02257424?term=NCT02257424&draw=2&rank=1 (accessed on 17 July 2020).
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, A.; Normandeau-Guimond, J.; Vaillancourt-Lavigueur, V.; Boivin, V.; Charbonneau, M.; Rivard, N.; Scott, M.S.; Dubois, C.M.; Jean, S. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci. Rep. 2019, 9, 11316. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellman, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J.-C. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Forman, H.J.; Ann, D.K. Novel roles for protein kinase Cdelta-dependent signaling pathways in acute hypoxic stress-induced autophagy. J. Biol. Chem. 2008, 283, 34432–34444. [Google Scholar] [CrossRef]
- Vasilevskaya, I.A.; Selvakumaran, M.; Roberts, D.; O’Dwyer, P.J. JNK1 Inhibition Attenuates Hypoxia-Induced Autophagy and Sensitizes to Chemotherapy. Mol. Cancer Res. 2016, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- FOLFOX/Bevacizumab/Hydroxychloroquine (HCQ) in Colorectal Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01206530?term=hydroxychloroquine&cond=colorectal+cancer&draw=2&rank=4 (accessed on 17 July 2020).
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.-M.; Li, B.; Guan, J.-J.; Xu, H.-D.; Shen, G.-H.; Gao, Q.-G.; Qin, Z.-H. Transcription factor EB is involved in autophagy-mediated chemoresistance to doxorubicin in human cancer cells. Acta Pharmacol. Sin. 2017, 38, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Thorburn, A. Targeting autophagy in breast cancer. World J. Clin. Oncol. 2014, 5, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2013, 44, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Keulers, T.G.; Schaaf, M.B.E.; Rouschop, K. Autophagy-Dependent Secretion: Contribution to Tumor Progression. Front. Oncol. 2016, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef]
- Lieblein, J.C.; Ball, S.; Hutzen, B.J.; Kate, S.A.; Lin, H.-J.L.; Huang, T.H.-M.; Hall, B.M.; Lin, L. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 2008, 8, 302. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Levy, J.M.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef]
- Janser, F.A.; Tschan, M.P.; Langer, R.; A Janser, F. The role of autophagy in HER2-targeted therapy. Swiss Med. Wkly. 2019, 149, w20138. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Feng, J.; Chang, Y.; Wang, Z.; Wen, A.-D. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med. Hypotheses 2011, 77, 206–208. [Google Scholar] [CrossRef]
- Aveic, S.; Tonini, G.P. Resistance to receptor tyrosine kinase inhibitors in solid tumors: Can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell Int. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Vázquez-Martín, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castilló, B.; Menendez, J.A. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: Utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012, 3, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.E.; Reidel, S.I.; Joffé, E.D.B.D.K.; Jasnis, M.A.; Fiszman, G.L. Autophagy Protects from Trastuzumab-Induced Cytotoxicity in HER2 Overexpressing Breast Tumor Spheroids. PLoS ONE 2015, 10, e0137920. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Martín, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy Facilitates the Development of Breast Cancer Resistance to the Anti-HER2 Monoclonal Antibody Trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; Lopez-Bonet, E.; Martin-Castilló, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes Primary resistance and restores sensitivity to Trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef]
- Janser, F.A.; Adams, O.; Bütler, V.; Schläfli, A.M.; Dislich, B.; Seiler, C.A.; Kröll, D.; Langer, R.; Tschan, M.P. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3069. [Google Scholar] [CrossRef]
- Negri, T.; Tarantino, E.; Orsenigo, M.; Reid, J.F.; Gariboldi, M.; Zambetti, M.; Pierotti, M.A.; Pilotti, S. Chromosome band 17q21 in breast cancer: Significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosom. Cancer 2010, 49, 901–909. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Lu, C.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. Interaction between Her2 and Beclin-1 Proteins Underlies a New Mechanism of Reciprocal Regulation. J. Boil. Chem. 2013, 288, 20315–20325. [Google Scholar] [CrossRef]
- Autophagy Inhibition Using Hydrochloroquine in Breast Cancer Patients. 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01292408?term=NCT01292408&draw=2&rank=1 (accessed on 17 July 2020).
- A Phase 2 Randomized, Double-blind Trial Evaluating the Effects of Chloroquine in Breast Cancer (CUBiC). 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02333890?term=NCT02333890&draw=2&rank=1 (accessed on 17 July 2020).
- Study of the Efficacy of Chloroquine in the Treatment of Ductal Carcinoma in Situ (The PINC Trial). 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT01023477?term=NCT01023477&draw=2&rank=1 (accessed on 17 July 2020).
- Chloroquine With Taxane Chemotherapy for Advanced or Metastatic Breast Cancer After Anthracycline Failure (CAT) (CAT). 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01446016?term=NCT01446016&draw=2&rank=1 (accessed on 17 July 2020).
- CLEVER Pilot Trial: A Phase II Pilot Trial of HydroxyChLoroquine, EVErolimus or the Combination for Prevention of Recurrent Breast Cancer. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03032406?term=NCT03032406&draw=2&rank=1 (accessed on 17 July 2020).
- Gedatolisib, Hydroxychloroquine or the Combination for Prevention of Recurrent Breast Cancer (“GLACIER”) (GLACIER). 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03400254?term=NCT03400254&draw=2&rank=1 (accessed on 17 July 2020).
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer. Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.D.; Rawson, J.V.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-Inhibition of BCL-W and BCL2 Restores Antiestrogen Sensitivity through BECN1 and Promotes an Autophagy-Associated Necrosis. PLoS ONE 2010, 5, e8604. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Koh, D.; Na, H.; Ka, N.-L.; Kim, S.; Kim, H.-J.; Hong, S.; Shin, Y.K.; Seong, J.K.; Lee, M.-O. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy 2018, 14, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, M.; Perna, E.; Tronnolone, S.; Colecchia, D.; Chiariello, M. Activated kinase screening identifies the IKBKE oncogene as a positive regulator of autophagy. Autophagy 2018, 15, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Long, Y.-H.; Wang, S.-Q.; Zhang, Y.-Y.; Li, Y.-F.; Mi, J.-S.; Yu, C.-H.; Li, D.-Y.; Zhang, J.-H.; Zhang, X.-J. JMJD6 regulates histone H2A.X phosphorylation and promotes autophagy in triple-negative breast cancer cells via a novel tyrosine kinase activity. Oncogene 2018, 38, 980–997. [Google Scholar] [CrossRef]
- Aveic, S.; Pantile, M.; Polo, P.; Sidarovich, V.; De Mariano, M.; Quattrone, A.; Longo, L.; Tonini, G.P. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hino, H.; Moriya, S.; Kazama, H.; Miyazaki, M.; Takano, N.; Hiramoto, M.; Tsukahara, K.; Miyazawa, K. Comparison of autophagy inducibility in various tyrosine kinase inhibitors and their enhanced cytotoxicity via inhibition of autophagy in cancer cells in combined treatment with azithromycin. Biochem. Biophys. Rep. 2020, 22, 100750. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR Tyrosine Kinase Inhibitors Activate Autophagy as a Cytoprotective Response in Human Lung Cancer Cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef]
- Kang, M.; Lee, K.-H.; Lee, H.S.; Jeong, C.W.; Kwak, C.; Kim, H.H.; Ku, J.H. Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 321. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [PubMed]
- Bokobza, S.M.; Jiang, Y.; Weber, A.M.; Devery, A.M.; Ryan, A.J. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget 2014, 5, 4765–4778. [Google Scholar] [CrossRef] [PubMed]
- Erlotinib With or Without Hydroxychloroquine in Chemo-Naive Advanced NSCLC and (EGFR) Mutations. 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT00977470?term=NCT00977470&draw=2&rank=1 (accessed on 17 July 2020).
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; White, E. Autophagy, metabolism, and cancer. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2016; Volume 81, pp. 73–78. [Google Scholar]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef]
- Huang, X.; Gan, G.; Wang, X.; Xu, T.; Xie, W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy 2019, 15, 1258–1279. [Google Scholar] [CrossRef]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Stockley, J.; Coffey, K.; O’Neill, D.; Jones, D.L.; Wade, M.; Wright, J.; Moore, M.; Tse, S.; Rogerson, L.; et al. KDM4B is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013, 41, 6892–6904. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. [Google Scholar] [CrossRef] [PubMed]
- Sha, J.; Han, Q.; Chi, C.; Zhu, Y.; Pan, J.; Dong, B.; Huang, Y.; Xia, W.; Xue, W. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J. Cell. Physiol. 2019, 235, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef]
- Jutten, B.; Keulers, T.G.; Schaaf, M.B.; Savelkouls, K.; Theys, J.; Span, P.N.; Vooijs, M.; Bussink, J.; Rouschop, K. EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival. Radiother. Oncol. 2013, 108, 479–483. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).