Revisiting the Concept of Stress in the Prognosis of Solid Tumors: A Role for Stress Granules Proteins?

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Generalities
2. SGs Are Composed of Proteins Involved in the Regulation of mRNA Translation
3. Stress Granules Are Pro-Survival Entities at the Cytoplasmic Level
- First, many pro-apoptotic signaling molecules are sequestered in SGs and it has been proposed that it prevents them from activating the pro-apoptotic cascade. This is the case for RACK1 (Receptor of Activated Protein C Kinase 1), TRAF2 (TNF receptor-associated factor 2), and RSK2 (Ribosomal S6 kinase 2) [45,46,47].
- Second, while not fully characterized, SGs protect cells from oxidative insults by reducing the level of cellular reactive oxygen species (ROS) [39,41,48]. Indeed, when the expression level of a major SGs regulator G3BP1 (RAS GTPase-activating Protein-Binding Protein 1) drops, ROS generation after exposure to oxidative insult increases. Moreover, the overexpression of G3BP1 reduces the level of ROS as compared to wild type cells. Cells expressing a truncated form of the protein that abrogates SGs formation have increased ROS production. Similar results have been obtained with USP10 (Ubiquitin carboxyl-terminal hydrolase 10), another SGs regulating protein [39].
- Third, SGs formation reduces the cellular energetic needs during stress by restricting the process of translation, which consumes a lot of ATP. By protecting mRNAs from stress-induced degradation, this allows cells to restart translation as soon as the stress is resolved without having to synthetize de novo RNAs [49]. Additionally, SGs sequester untranslated mRNAs concomitantly with the global inhibition of translation [17]. Some mRNAs, such as chaperone mRNAs, are excluded from the SGs structures, so that they can be preferentially translated during the time of the stress and participate in the proper protein folding and avoid functional defects [17,19,50]. By these actions, SGs are described as triage centers for translation of mRNAs during stress exposure [17]. One leading hypothesis is that SGs are able to reshape translational patterns under stress exposure [19,51].
4. Stress Granules Are Involved in Cancer Progression
5. SGs Regulators and Their Role in Cancer
6. SGs Proteins as Prognostic Markers: At the mRNA or Protein Level?
7. Conclusions
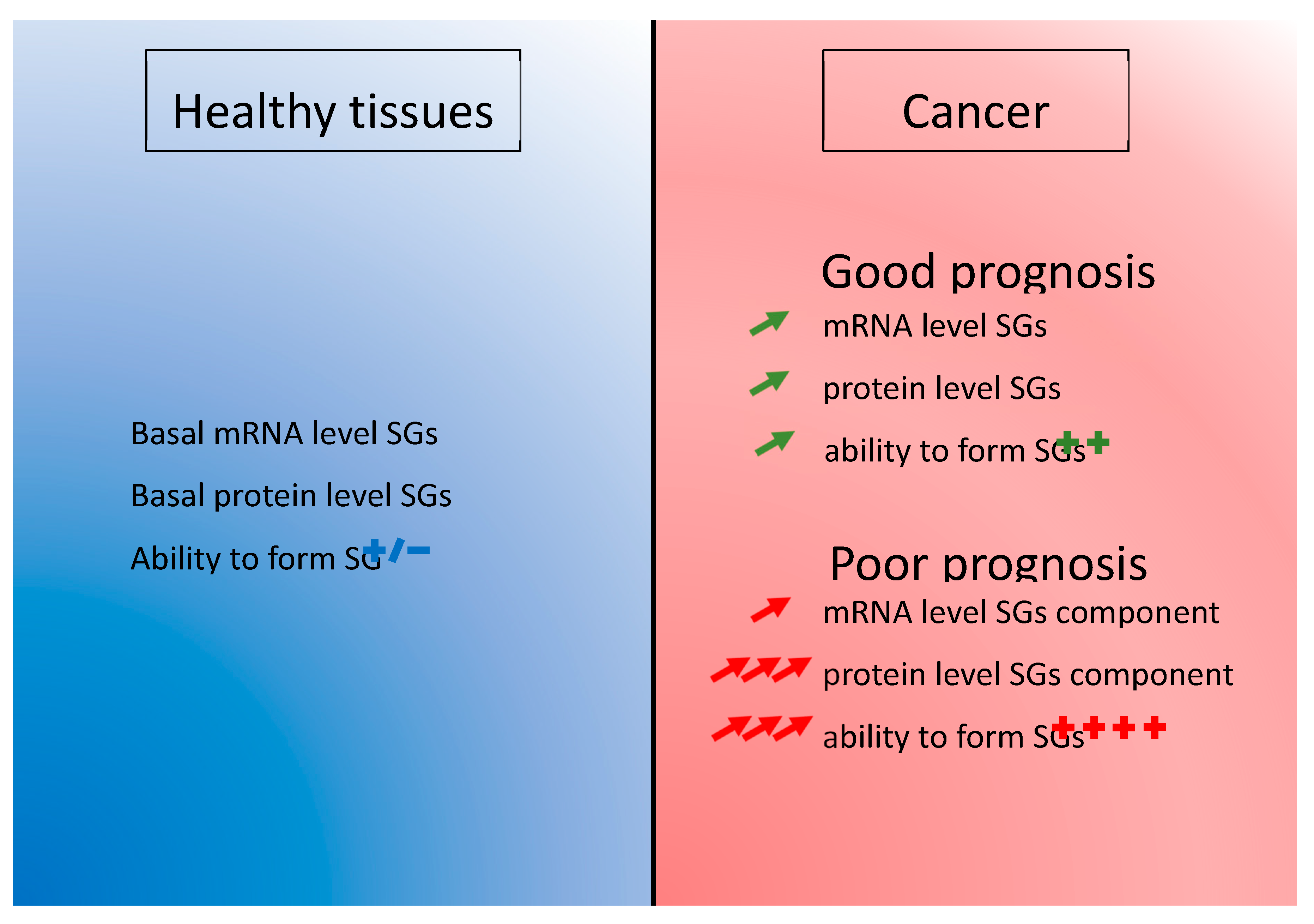
- An early increased of mRNA level in the pathology achieved by upregulation of transcription or decrease of RNA degradation. This phenomenon could insure an efficient level of the proteins that are involved in SGs function. This basal level is potentially the result of cancer cells subversion of the SGs mechanism to ensure their survival and it is present in the majority of cancer patients (Figure 4).
- A post-transcriptional regulation impacting the final level of proteins. Increasing the translation rate of the mRNA encoding a SGs protein would be more efficient than producing more mRNAs. This could be done by specific signal in the non-translated region of the mRNA such as AU-rich element (ARE) [153] or to the length of the mRNA as it was already described [154]. Favoring translation over transcription is perhaps also a mechanism allowing an economy of energy in cancer cells (Figure 4).
- Or a post-translational modification enhancing the half-life of the protein. The degradation of the protein is delayed and it enhances the overall level of protein (Figure 4).
8. Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Norouzi, S.; Gorgi Valokala, M.; Mosaffa, F.; Zirak, M.R.; Zamani, P.; Behravan, J. Crosstalk in cancer resistance and metastasis. Crit. Rev. Oncol. Hematol. 2018, 132, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-Jones, S.; Zhou, X.; Cai, S.; Tu, Y.; McCoy, A.; Peoples, M.; et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J.Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [Green Version]
- Aulas, A.; Fay, M.M.; Lyons, S.M.; Achorn, C.A.; Kedersha, N.; Anderson, P.; Ivanov, P. Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Kaehler, C.; Isensee, J.; Hucho, T.; Lehrach, H.; Krobitsch, S. 5-Fluorouracil affects assembly of stress granules based on RNA incorporation. Nucl. Acids Res. 2014, 42, 6436–6447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, K.; Sasaki, A.T.; Anderson, P. Selenite targets eIF4E-binding protein-1 to inhibit translation initiation and induce the assembly of non-canonical stress granules. Nucl. Acids Res. 2012, 40, 8099–8110. [Google Scholar] [CrossRef] [PubMed]
- Adjibade, P.; St-Sauveur, V.G.; Quevillon Huberdeau, M.; Fournier, M.J.; Savard, A.; Coudert, L.; Khandjian, E.W.; Mazroui, R. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget 2015, 6, 43927–43943. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.J.; Gareau, C.; Mazroui, R. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer Cell Int. 2010, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaflarski, W.; Fay, M.M.; Kedersha, N.; Zabel, M.; Anderson, P.; Ivanov, P. Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 2016, 7, 30307–30322. [Google Scholar] [CrossRef]
- Vilas-Boas Fde, A.; da Silva, A.M.; de Sousa, L.P.; Lima, K.M.; Vago, J.P.; Bittencourt, L.F.; Dantas, A.E.; Gomes, D.A.; Vilela, M.C.; Teixeira, M.M.; et al. Impairment of stress granule assembly via inhibition of the eIF2alpha phosphorylation sensitizes glioma cells to chemotherapeutic agents. J. Neurooncol. 2016, 127, 253–260. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [Green Version]
- Timalsina, S.; Arimoto-Matsuzaki, K.; Kitamura, M.; Xu, X.; Wenzhe, Q.; Ishigami-Yuasa, M.; Kagechika, H.; Hata, Y. Chemical compounds that suppress hypoxia-induced stress granule formation enhance cancer drug sensitivity of human cervical cancer HeLa cells. J. Biochem. 2018, 164, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Somasekharan, S.P.; Zhang, F.; Saxena, N.; Huang, J.N.; Kuo, I.C.; Low, C.; Bell, R.; Adomat, H.; Stoynov, N.; Foster, L.; et al. G3BP1-linked mRNA partitioning supports selective protein synthesis in response to oxidative stress. Nucl. Acids Res. 2020. [Google Scholar] [CrossRef]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Grabocka, E. Stress Granules in Cancer. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Nover, L.; Scharf, K.D.; Neumann, D. Formation of cytoplasmic heat shock granules in tomato cell cultures and leaves. Mol. Cell. Biol. 1983, 3, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiardi, D.A.; McLaughlin-Williamson, K.; May, K.E.; Messana, E.P.; Mountain, D.C.; Cotanche, D.A. Progression of hair cell ejection and molecular markers of apoptosis in the avian cochlea following gentamicin treatment. J. Comp. Neurol. 2004, 475, 1–18. [Google Scholar] [CrossRef]
- Anderson, E.N.; Gochenaur, L.; Singh, A.; Grant, R.; Patel, K.; Watkins, S.; Wu, J.Y.; Pandey, U.B. Traumatic injury induces Stress Granule Formation and enhances Motor Dysfunctions in ALS/FTD Models. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Scharf, K.D.; Heider, H.; Hohfeld, I.; Lyck, R.; Schmidt, E.; Nover, L. The tomato Hsf system: HsfA2 needs interaction with HsfA1 for efficient nuclear import and may be localized in cytoplasmic heat stress granules. Mol. Cell Biol. 1998, 18, 2240–2251. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Kwon, S.H.; Su, Y.; Dong, Z. Stress granules are formed in renal proximal tubular cells during metabolic stress and ischemic injury for cell survival. Am. J. Physiol. Ren. Physiol. 2019, 317, F116–F123. [Google Scholar] [CrossRef]
- Souquere, S.; Mollet, S.; Kress, M.; Dautry, F.; Pierron, G.; Weil, D. Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J. Cell Sci. 2009, 122, 3619–3626. [Google Scholar] [CrossRef] [Green Version]
- Aulas, A.; Vande Velde, C. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, J.R.; Jain, S.; Khong, A.; Parker, R. Isolation of yeast and mammalian stress granule cores. Methods 2017, 126, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Adivarahan, S.; Livingston, N.; Nicholson, B.; Rahman, S.; Wu, B.; Rissland, O.S.; Zenklusen, D. Spatial Organization of Single mRNPs at Different Stages of the Gene Expression Pathway. Mol. Cell 2018, 72, 727–738.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Kedersha, N.; Low, W.K.; Romo, D.; Gorospe, M.; Kaufman, R.; Anderson, P.; Liu, J.O. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J. Biol. Chem. 2006, 281, 32870–32878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulas, A.; Lyons, S.M.; Fay, M.M.; Anderson, P.; Ivanov, P. Nitric oxide triggers the assembly of “type II” stress granules linked to decreased cell viability. Cell Death Dis. 2018, 9, 1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, N.; Kunzli, C.; Buthey, K.; Saxena, S. C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Mol. Neurobiol. 2017, 54, 3062–3077. [Google Scholar] [CrossRef]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orru, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 2016, 25, 4473–4483. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Stabile, S.; Vande Velde, C. Endogenous TDP-43, but not FUS, contributes to stress granule assembly via G3BP. Mol. Neurodegener. 2012, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulas, A.; Stabile, S.; Vande Velde, C. Erratum to: Endogenous TDP-43, but not FUS, contributes to stress granule assembly via G3BP. Mol. Neurodegener. 2015, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger-Mathason, T.S.; Andrade, J.; Groehler, A.L.; Clark, D.E.; Muratore-Schroeder, T.L.; Pasic, L.; Smith, J.A.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G.; et al. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol. Cell 2008, 31, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Kim, W.J.; Back, S.H.; Kim, V.; Ryu, I.; Jang, S.K. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cellul. Biol. 2005, 25, 2450–2462. [Google Scholar] [CrossRef] [Green Version]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Aulas, A.; Caron, G.; Gkogkas, C.G.; Mohamed, N.V.; Destroismaisons, L.; Sonenberg, N.; Leclerc, N.; Parker, J.A.; Vande Velde, C. G3BP1 promotes stress-induced RNA granule interactions to preserve polyadenylated mRNA. J. Cell Biol. 2015, 209, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Gareau, C.; Fournier, M.J.; Filion, C.; Coudert, L.; Martel, D.; Labelle, Y.; Mazroui, R. p21(WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS ONE 2011, 6, e20254. [Google Scholar] [CrossRef]
- Goncalves, A.C.; Towers, E.R.; Haq, N.; Porco, J.A., Jr.; Pelletier, J.; Dawson, S.J.; Gale, J.E. Drug-induced Stress Granule Formation Protects Sensory Hair Cells in Mouse Cochlear Explants During Ototoxicity. Sci. Rep. 2019, 9, 12501. [Google Scholar] [CrossRef]
- Adjibade, P.; Mazroui, R. Control of mRNA turnover: Implication of cytoplasmic RNA granules. Semin. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.G.; Conn, C.S.; Kye, Y.; Xue, L.; Forester, C.M.; Cowan, J.E.; Hsieh, A.C.; Cunningham, J.T.; Truillet, C.; Tameire, F.; et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Taylor, M.J.; Walsh, L.A.; Dieters-Castator, D.; Das, P.; Jewer, M.; Zhang, G.; Postovit, L.M. Low oxygen levels induce the expression of the embryonic morphogen Nodal. Mol. Biol. Cell 2011, 22, 4809–4821. [Google Scholar] [CrossRef]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewer, M.L.; Zhang, G.; Liu, J.; Findlay, S.; Vincent, K.; Tandoc, K.; Dieters-Castator, D.; Quail, D.; Dutta, I.; Coatham, M.; et al. Translational control of breast cancer plasticity. BioRxviv 2019. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The TIA1 RNA-Binding Protein Family Regulates EIF2AK2-Mediated Stress Response and Cell Cycle Progression. Mol. Cell 2018, 69, 622–635.e6. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Jimenez, C.; Ludena, M.D.; Izquierdo, J.M. T-cell intracellular antigens function as tumor suppressor genes. Cell Death Dis. 2015, 6, e1669. [Google Scholar] [CrossRef]
- Lafarga, V.; Sung, H.M.; Haneke, K.; Roessig, L.; Pauleau, A.L.; Bruer, M.; Rodriguez-Acebes, S.; Lopez-Contreras, A.J.; Gruss, O.J.; Erhardt, S.; et al. TIAR marks nuclear G2/M transition granules and restricts CDK1 activity under replication stress. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Hamdollah Zadeh, M.A.; Amin, E.M.; Hoareau-Aveilla, C.; Domingo, E.; Symonds, K.E.; Ye, X.; Heesom, K.J.; Salmon, A.; D’Silva, O.; Betteridge, K.B.; et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol. Oncol. 2015, 9, 167–178. [Google Scholar] [CrossRef]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef]
- Gupta, N.; Badeaux, M.; Liu, Y.; Naxerova, K.; Sgroi, D.; Munn, L.L.; Jain, R.K.; Garkavtsev, I. Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc. Natl. Acad. Sci. USA 2017, 114, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitard, E.; Parker, F.; Millon, R.; Abecassis, J.; Tocque, B. G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett. 2001, 162, 213–221. [Google Scholar] [CrossRef]
- Taniuchi, K.; Nishimori, I.; Hollingsworth, M.A. The N-terminal domain of G3BP enhances cell motility and invasion by posttranscriptional regulation of BART. Mol. Cancer Res. MCR 2011, 9, 856–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fu, D.; Chen, Y.; Su, J.; Wang, Y.; Li, X.; Zhai, W.; Niu, Y.; Yue, D.; Geng, H. G3BP1 promotes tumor progression and metastasis through IL-6/G3BP1/STAT3 signaling axis in renal cell carcinomas. Cell Death Dis. 2018, 9, 501. [Google Scholar] [CrossRef] [PubMed]
- Dou, N.; Chen, J.; Yu, S.; Gao, Y.; Li, Y. G3BP1 contributes to tumor metastasis via upregulation of Slug expression in hepatocellular carcinoma. Am. J. Cancer Res. 2016, 6, 2641–2650. [Google Scholar]
- Cho, E.; Than, T.T.; Kim, S.H.; Park, E.R.; Kim, M.Y.; Lee, K.H.; Shin, H.J. G3BP1 Depletion Increases Radiosensitisation by Inducing Oxidative Stress in Response to DNA Damage. Anticancer. Res. 2019, 39, 6087–6095. [Google Scholar] [CrossRef]
- Heijink, A.M.; Everts, M.; Honeywell, M.E.; Richards, R.; Kok, Y.P.; de Vries, E.G.E.; Lee, M.J.; van Vugt, M. Modeling of Cisplatin-Induced Signaling Dynamics in Triple-Negative Breast Cancer Cells Reveals Mediators of Sensitivity. Cell Rep. 2019, 28, 2345–2357.e5. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; David, M.D.; Schrader, J.W. Absence of caprin-1 results in defects in cellular proliferation. J. Immunol. 2005, 175, 4274–4282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Hu, H.; Chen, J.; Cao, S.; Yu, J.; Xue, J.; Chen, F.; Cai, Y.; He, H.; Zhang, L. Caprin-1 is a novel microRNA-223 target for regulating the proliferation and invasion of human breast cancer cells. Biomed. Pharmacother. 2013, 67, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.S.; Qing, H.; Gui, H.; Luo, J.; Dai, L.J.; Wang, B. Role of caprin-1 in carcinogenesis. Oncol. Lett. 2019, 18, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Li, T.; Zhao, Y.; Huang, L.; Sun, H.; Wu, H.; Jiang, X. USP10 inhibits lung cancer cell growth and invasion by upregulating PTEN. Mol. Cell Biochem. 2018, 441, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ning, Z.; Wang, A.; Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Li, T.; Liu, X.; et al. USP10 suppresses tumor progression by inhibiting mTOR activation in hepatocellular carcinoma. Cancer lett. 2018, 436, 139–148. [Google Scholar] [CrossRef]
- Lee, A.K.; Klein, J.; Fon Tacer, K.; Lord, T.; Oatley, M.J.; Oatley, J.M.; Porter, S.N.; Pruett-Miller, S.M.; Tikhonova, E.B.; Karamyshev, A.L.; et al. Translational Repression of G3BP in Cancer and Germ Cells Suppresses Stress Granules and Enhances Stress Tolerance. Mol. Cell 2020. [Google Scholar] [CrossRef]
- van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [Green Version]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Hess, K.R.; Anderson, K.; Symmans, W.F.; Valero, V.; Ibrahim, N.; Mejia, J.A.; Booser, D.; Theriault, R.L.; Buzdar, A.U.; Dempsey, P.J.; et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J. Clin. Oncol. 2006, 24, 4236–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivshina, A.V.; George, J.; Senko, O.; Mow, B.; Putti, T.C.; Smeds, J.; Lindahl, T.; Pawitan, Y.; Hall, P.; Nordgren, H.; et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66, 10292–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoi, H.; Potti, A.; Delorenzi, M.; Mauriac, L.; Campone, M.; Tubiana-Hulin, M.; Petit, T.; Rouanet, P.; Jassem, J.; Blot, E.; et al. Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: A substudy of the EORTC 10994/BIG 00-01 clinical trial. Lancet Oncol. 2007, 8, 1071–1078. [Google Scholar] [CrossRef]
- Desmedt, C.; Piette, F.; Loi, S.; Wang, Y.; Lallemand, F.; Haibe-Kains, B.; Viale, G.; Delorenzi, M.; Zhang, Y.; d’Assignies, M.S.; et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res. 2007, 13, 3207–3214. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Larionov, A. Changes in expression of oestrogen regulated and proliferation genes with neoadjuvant treatment highlight heterogeneity of clinical resistance to the aromatase inhibitor, letrozole. Breast Cancer Res. 2010, 12, R52. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.; Wessel, R.; Graessmann, M.; Jurgens, M.; Petersen, I.; Schmutzler, R.; Niederacher, D.; Arnold, N.; Meindl, A.; Scherneck, S.; et al. Comparison of gene expression data from human and mouse breast cancers: Identification of a conserved breast tumor gene set. Int. J. Cancer 2007, 121, 683–688. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [Green Version]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdes, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Bohm, D.; von Torne, C.; Steiner, E.; Puhl, A.; Pilch, H.; Lehr, H.A.; Hengstler, J.G.; Kolbl, H.; Gehrmann, M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008, 68, 5405–5413. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Ganesan, K.; Tan, L.K.; Laban, M.; Wu, J.; Zhao, X.D.; Li, H.; Leung, C.H.; Zhu, Y.; Wei, C.L.; et al. A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, W.T.; Kernagis, D.N.; Dressman, H.K.; Griffis, R.J.; Hunter, J.D.; Olson, J.A.; Marks, J.R.; Ginsburg, G.S.; Marcom, P.K.; Nevins, J.R.; et al. Intratumor heterogeneity and precision of microarray-based predictors of breast cancer biology and clinical outcome. J. Clin. Oncol. 2010, 28, 2198–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Bianchini, G.; Booser, D.; Qi, Y.; Coutant, C.; Shiang, C.Y.; Santarpia, L.; Matsuoka, J.; Hortobagyi, G.N.; Symmans, W.F.; et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J. Natl. Cancer Inst. 2011, 103, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Korde, L.A.; Lusa, L.; McShane, L.; Lebowitz, P.F.; Lukes, L.; Camphausen, K.; Parker, J.S.; Swain, S.M.; Hunter, K.; Zujewski, J.A. Gene expression pathway analysis to predict response to neoadjuvant docetaxel and capecitabine for breast cancer. Breast Cancer Res. Treat. 2010, 119, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Tabchy, A.; Valero, V.; Vidaurre, T.; Lluch, A.; Gomez, H.; Martin, M.; Qi, Y.; Barajas-Figueroa, L.J.; Souchon, E.; Coutant, C.; et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin. Cancer Res. 2010, 16, 5351–5361. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, S.; Olsson, B.; Jacobsson, S.; Palmqvist, L.; Ricksten, A.; Ekeland-Sjoberg, K.; Wadenvik, H. BCR-ABL1 transcript levels increase in peripheral blood but not in granulocytes after physical exercise in patients with chronic myeloid leukemia. Scand. J. Clin. Lab. Investig. 2011, 71, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Di Leo, A.; de Azambuja, E.; Larsimont, D.; Haibe-Kains, B.; Selleslags, J.; Delaloge, S.; Duhem, C.; Kains, J.P.; Carly, B.; et al. Multifactorial approach to predicting resistance to anthracyclines. J. Clin. Oncol. 2011, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Guedj, M.; Marisa, L.; de Reynies, A.; Orsetti, B.; Schiappa, R.; Bibeau, F.; MacGrogan, G.; Lerebours, F.; Finetti, P.; Longy, M.; et al. A refined molecular taxonomy of breast cancer. Oncogene 2012, 31, 1196–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici, V.; Chen, W.; Gallas, B.G.; Hatzis, C.; Shi, W.; Samuelson, F.W.; Nikolsky, Y.; Tsyganova, M.; Ishkin, A.; Nikolskaya, T.; et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010, 12, R5. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Sabatier, R.; Finetti, P.; Adelaide, J.; Guille, A.; Borg, J.P.; Chaffanet, M.; Lane, L.; Birnbaum, D.; Bertucci, F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS ONE 2011, 6, e27656. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sieuwerts, A.M.; McGreevy, M.; Casey, G.; Cufer, T.; Paradiso, A.; Harbeck, N.; Span, P.N.; Hicks, D.G.; Crowe, J.; et al. The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast Cancer Res. Treat. 2009, 116, 303–309. [Google Scholar] [CrossRef]
- Chen, D.T.; Nasir, A.; Culhane, A.; Venkataramu, C.; Fulp, W.; Rubio, R.; Wang, T.; Agrawal, D.; McCarthy, S.M.; Gruidl, M.; et al. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res. Treat. 2010, 119, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorissen, R.N.; Gibbs, P.; Christie, M.; Prakash, S.; Lipton, L.; Desai, J.; Kerr, D.; Aaltonen, L.A.; Arango, D.; Kruhoffer, M.; et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7642–7651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffer, M.; Bacolod, M.D.; Zuk, O.; Giardina, S.F.; Pincas, H.; Barany, F.; Paty, P.B.; Gerald, W.L.; Notterman, D.A.; Domany, E. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7131–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staub, E.; Groene, J.; Heinze, M.; Mennerich, D.; Roepcke, S.; Klaman, I.; Hinzmann, B.; Castanos-Velez, E.; Pilarsky, C.; Mann, B.; et al. An expression module of WIPF1-coexpressed genes identifies patients with favorable prognosis in three tumor types. J. Mol. Med. 2009, 87, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Deane, N.G.; Wu, F.; Merchant, N.B.; Zhang, B.; Jiang, A.; Lu, P.; Johnson, J.C.; Schmidt, C.; Bailey, C.E.; et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 2010, 138, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, R.D.; Bylesjo, M.; Kerr, P.; Davison, T.; Black, J.M.; Kay, E.W.; Holt, R.J.; Proutski, V.; Ahdesmaki, M.; Farztdinov, V.; et al. Development and independent validation of a prognostic assay for stage II colon cancer using formalin-fixed paraffin-embedded tissue. J. Clin. Oncol. 2011, 29, 4620–4626. [Google Scholar] [CrossRef] [Green Version]
- Sveen, A.; Agesen, T.H.; Nesbakken, A.; Rognum, T.O.; Lothe, R.A.; Skotheim, R.I. Transcriptome instability in colorectal cancer identified by exon microarray analyses: Associations with splicing factor expression levels and patient survival. Genome Med. 2011, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Laibe, S.; Lagarde, A.; Ferrari, A.; Monges, G.; Birnbaum, D.; Olschwang, S.; Project, C.O.L. A seven-gene signature aggregates a subgroup of stage II colon cancers with stage III. OMICS 2012, 16, 560–565. [Google Scholar] [CrossRef]
- Marisa, L.; de Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Badea, R.; Socaciu, M.; Lupsor, M.; Mosteanu, O.; Pop, T. Evaluating the liver tumors using three-dimensional ultrasonography. A pictorial essay. J. Gastrointestin Liver Dis. 2007, 16, 85–92. [Google Scholar] [PubMed]
- Van den Broeck, A.; Vankelecom, H.; Van Eijsden, R.; Govaere, O.; Topal, B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2012, 31, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunardi, S.; Jamieson, N.B.; Lim, S.Y.; Griffiths, K.L.; Carvalho-Gaspar, M.; Al-Assar, O.; Yameen, S.; Carter, R.C.; McKay, C.J.; Spoletini, G.; et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget 2014, 5, 11064–11080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Kim, M.; Hwang, D.; Park, M.; Kim, W.K.; Kim, S.K.; Shin, J.; Park, E.S.; Kang, C.M.; Paik, Y.K.; et al. Characterization of gene expression and activated signaling pathways in solid-pseudopapillary neoplasm of pancreas. Mod. Pathol. 2014, 27, 580–593. [Google Scholar] [CrossRef] [Green Version]
- Grutzmann, R.; Saeger, H.D.; Luttges, J.; Schackert, H.K.; Kalthoff, H.; Kloppel, G.; Pilarsky, C. Microarray-based gene expression profiling in pancreatic ductal carcinoma: Status quo and perspectives. Int. J. Colorectal Dis. 2004, 19, 401–413. [Google Scholar] [CrossRef]
- Monzon, F.A.; Lyons-Weiler, M.; Buturovic, L.J.; Rigl, C.T.; Henner, W.D.; Sciulli, C.; Dumur, C.I.; Medeiros, F.; Anderson, G.G. Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. J. Clin. Oncol. 2009, 27, 2503–2508. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Kirby, M.K.; Ramaker, R.C.; Gertz, J.; Davis, N.S.; Johnston, B.E.; Oliver, P.G.; Sexton, K.C.; Greeno, E.W.; Christein, J.D.; Heslin, M.J.; et al. RNA sequencing of pancreatic adenocarcinoma tumors yields novel expression patterns associated with long-term survival and reveals a role for ANGPTL4. Mol. Oncol. 2016, 10, 1169–1182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Schetter, A.; He, P.; Funamizu, N.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Hassan, R.; Yfantis, H.G.; Lee, D.H.; et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e31507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, C.; Kristiansen, G.; Kersting, S.; Roy, J.; Aust, D.; Knosel, T.; Rummele, P.; Jahnke, B.; Hentrich, V.; Ruckert, F.; et al. Google goes cancer: Improving outcome prediction for cancer patients by network-based ranking of marker genes. PLoS Comput. Biol. 2012, 8, e1002511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Stratford, J.K.; Bentrem, D.J.; Anderson, J.M.; Fan, C.; Volmar, K.A.; Marron, J.S.; Routh, E.D.; Caskey, L.S.; Samuel, J.C.; Der, C.J.; et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010, 7, e1000307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.T.; Davis-Yadley, A.H.; Huang, P.Y.; Husain, K.; Centeno, B.A.; Permuth-Wey, J.; Pimiento, J.M.; Malafa, M. Prognostic Fifteen-Gene Signature for Early Stage Pancreatic Ductal Adenocarcinoma. PLoS ONE 2015, 10, e0133562. [Google Scholar] [CrossRef]
- French, J.; Stirling, R.; Walsh, M.; Kennedy, H.D. The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem. J. 2002, 34, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Somasekharan, S.P.; El-Naggar, A.; Leprivier, G.; Cheng, H.; Hajee, S.; Grunewald, T.G.; Zhang, F.; Ng, T.; Delattre, O.; Evdokimova, V.; et al. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J. Cell Biol. 2015, 208, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhan, Y.; Zhang, Y.; Liu, S.; Lu, J.; Yang, Y.; Wen, Q.; Fan, S. Elevated expression of G3BP1 associates with YB1 and p-AKT and predicts poor prognosis in nonsmall cell lung cancer patients after surgical resection. Cancer Med. 2019, 8, 6894–6903. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; Ruan, Y.; Shen, Z.; Jia, D.; Wang, X.; Zhao, J.; Sun, Y.; Gu, J. Overexpression of Ras-GTPase-activating protein SH3 domain-binding protein 1 correlates with poor prognosis in gastric cancer patients. Histopathology 2015, 67, 677–688. [Google Scholar] [CrossRef]
- Xiong, R.; Gao, J.L.; Yin, T. G3BP1 activates the TGF-beta/Smad signaling pathway to promote gastric cancer. Onco Targets Ther. 2019, 12, 7149–7156. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.I.; Suzuki, T.; Fujimura, T.; Takahashi, S.; Inoue, S. Association of USP10 with G3BP2 Inhibits p53 Signaling and Contributes to Poor Outcome in Prostate Cancer. Mol. Cancer Res. MCR 2018, 16, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlobec, I.; Karamitopoulou, E.; Terracciano, L.; Piscuoglio, S.; Iezzi, G.; Muraro, M.G.; Spagnoli, G.; Baker, K.; Tzankov, A.; Lugli, A. TIA-1 cytotoxic granule-associated RNA binding protein improves the prognostic performance of CD8 in mismatch repair-proficient colorectal cancer. PLoS ONE 2010, 5, e14282. [Google Scholar] [CrossRef] [PubMed]
- Hasselblom, S.; Sigurdadottir, M.; Hansson, U.; Nilsson-Ehle, H.; Ridell, B.; Andersson, P.O. The number of tumour-infiltrating TIA-1+ cytotoxic T cells but not FOXP3+ regulatory T cells predicts outcome in diffuse large B-cell lymphoma. Br. J. Haematol. 2007, 137, 364–373. [Google Scholar] [CrossRef]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Sabile, A.A.; Arlt, M.J.; Muff, R.; Husmann, K.; Hess, D.; Bertz, J.; Langsam, B.; Aemisegger, C.; Ziegler, U.; Born, W.; et al. Caprin-1, a novel Cyr61-interacting protein, promotes osteosarcoma tumor growth and lung metastasis in mice. Biochim. Biophys. Acta 2013, 1832, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Tan, N.; Dai, L.; Liu, X.; Pan, G.; Chen, H.; Huang, J.; Xu, Q. Upregulation of caprin1 expression is associated with poor prognosis in hepatocellular carcinoma. Pathol. Res. Pract. 2017, 213, 1563–1567. [Google Scholar] [CrossRef]
- Zhang, Y.; You, W.; Zhou, H.; Chen, Z.; Han, G.; Zuo, X.; Zhang, L.; Wu, J.; Wang, X. Downregulated miR-621 promotes cell proliferation via targeting CAPRIN1 in hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 2116–2129. [Google Scholar]
- Zeng, Z.; Wu, H.X.; Zhan, N.; Huang, Y.B.; Wang, Z.S.; Yang, G.F.; Wang, P.; Fu, G.H. Prognostic significance of USP10 as a tumor-associated marker in gastric carcinoma. Tumour Biol. 2014, 35, 3845–3853. [Google Scholar] [CrossRef]
- Han, G.H.; Chay, D.B.; Yi, J.M.; Cho, H.; Chung, J.Y.; Kim, J.H. Loss of Both USP10 and p14ARF Protein Expression Is an Independent Prognostic Biomarker for Poor Prognosis in Patients With Epithelial Ovarian Cancer. Cancer Genom. Proteom. 2019, 16, 553–562. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, D.; Yu, T.; Huang, Y.; Yan, H.; Gu, L.; Yuan, J. Association and clinical implication of the USP10 and MSH2 proteins in non-small cell lung cancer. Oncol. Lett. 2019, 17, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.S.; Yi, J.M.; Cho, H.; Choi, C.H.; Park, Y.; Chung, E.J.; Song, J.; Chung, J.Y.; Hong, S.M. Dual loss of USP10 and p14ARF protein expression is associated with poor prognosis in patients with small intestinal adenocarcinoma. Tumour Biol. 2018, 40, 1010428318808678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, H.; Fukao, A.; Funakami, Y.; Duncan, K.E.; Fujiwara, T. Emerging Evidence of Translational Control by AU-Rich Element-Binding Proteins. Front. Genet. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820.e5. [Google Scholar] [CrossRef]
- Dai, L.; Lin, Z.; Cao, Y.; Chen, Y.; Xu, Z.; Qin, Z. Targeting EIF4F complex in non-small cell lung cancer cells. Oncotarget 2017, 8, 55731–55735. [Google Scholar] [CrossRef]
- Chan, K.; Robert, F.; Oertlin, C.; Kapeller-Libermann, D.; Avizonis, D.; Gutierrez, J.; Handly-Santana, A.; Doubrovin, M.; Park, J.; Schoepfer, C.; et al. eIF4A supports an oncogenic translation program in pancreatic ductal adenocarcinoma. Nat. Commun. 2019, 10, 5151. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; Rong, L.; Yoshida, K.; Ler, L.W.; Sonenberg, N. eIF4E--from translation to transformation. Oncogene 2004, 23, 3172–3179. [Google Scholar] [CrossRef] [Green Version]
- Hershey, J.W. The role of eIF3 and its individual subunits in cancer. Biochim. Biophys. Acta 2015, 1849, 792–800. [Google Scholar] [CrossRef]
- Yin, Y.; Long, J.; Sun, Y.; Li, H.; Jiang, E.; Zeng, C.; Zhu, W. The function and clinical significance of eIF3 in cancer. Gene 2018, 673, 130–133. [Google Scholar] [CrossRef]
- Yoshida, K.; Kajiyama, H.; Inami, E.; Tamauchi, S.; Ikeda, Y.; Yoshikawa, N.; Nishino, K.; Utsumi, F.; Niimi, K.; Suzuki, S.; et al. Clinical Significance of Ubiquitin-associated Protein 2-like in Patients With Uterine Cervical Cancer. In Vivo 2020, 34, 109–116. [Google Scholar] [CrossRef]
- Frydryskova, K.; Masek, T.; Borcin, K.; Mrvova, S.; Venturi, V.; Pospisek, M. Distinct recruitment of human eIF4E isoforms to processing bodies and stress granules. BMC Mol. Biol. 2016, 17, 21. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, L.; Cieren, A.; Barbieri, S.; Khong, A.; Schwager, F.; Parker, R.; Gotta, M. UBAP2L Forms Distinct Cores that Act in Nucleating Stress Granules Upstream of G3BP1. Curr. Biol. 2020, 30, 698–707.e6. [Google Scholar] [CrossRef] [PubMed]
- Palam, L.R.; Gore, J.; Craven, K.E.; Wilson, J.L.; Korc, M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015, 6, e1913. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Yang, P.; Zhang, Y.; Bao, X.; Li, J.; Hou, W.; Yao, X.; Han, J.; Zhang, H. A genome-wide RNAi screen identifies genes regulating the formation of P bodies in C. elegans and their functions in NMD and RNAi. Protein Cell 2011, 2, 918–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabocka, E.; Bar-Sagi, D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016, 167, 1803–1813.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

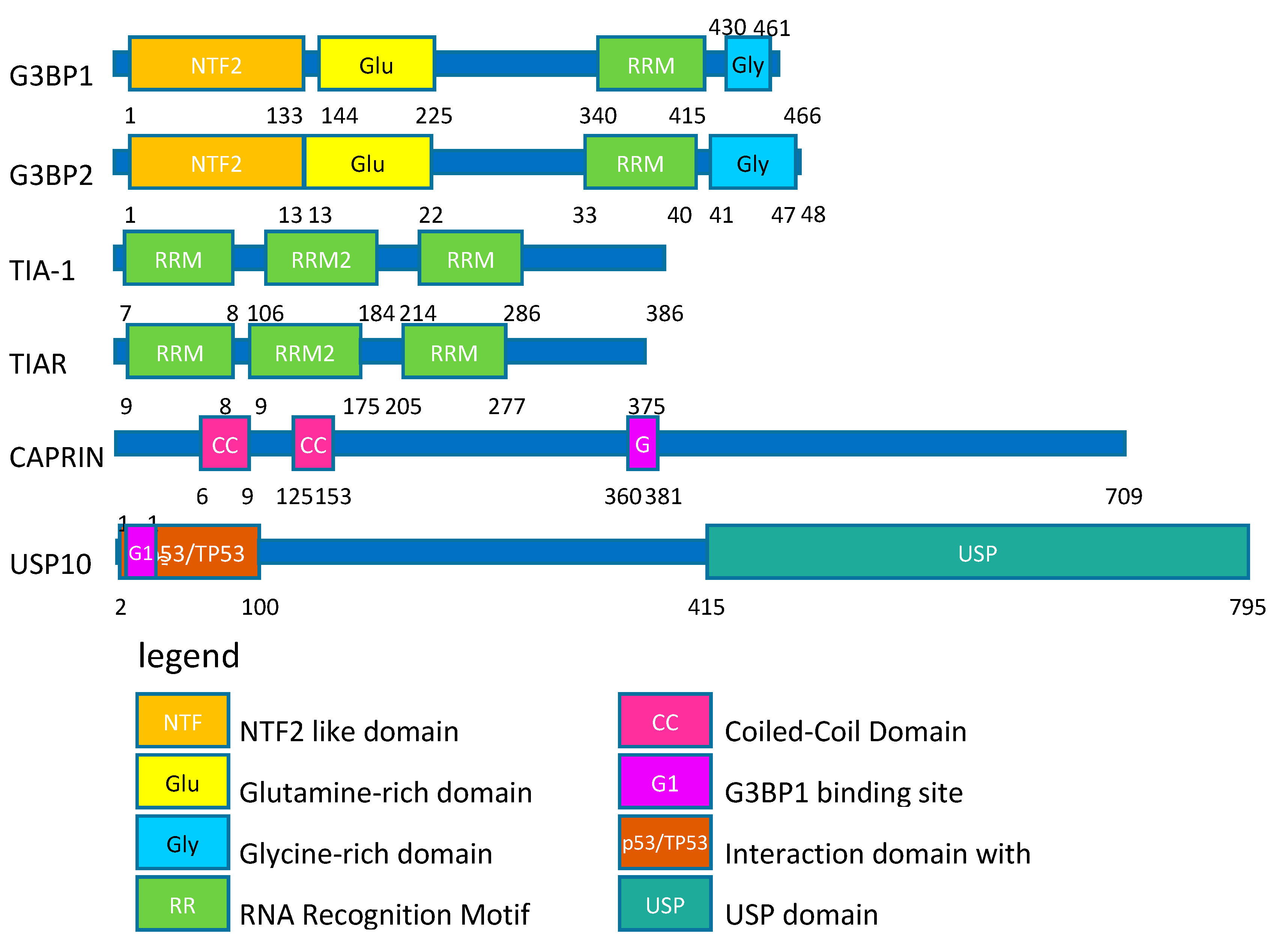



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | G3BP1 | G3BP2 | TIA-1 | CAPRIN-1 | USP10 |
|---|---|---|---|---|---|
| Breast | Poor [136] | Poor [137] | |||
| Colon/Colo-Rectal | Poor [143] | ||||
| Sarcoma | Poor [138] | ||||
| Stomach | Poor [140,141] | Good [149] | |||
| Lung | Poor [139] | Good [151] | |||
| Liver | Poor [68] | Poor [145] | Poor [147,148] | ||
| Prostate | Poor [142] | Good [142] | |||
| Intestinal | Good [152] | ||||
| Ovarian | Good [150] | ||||
| Osteosarcoma | Poor [146] | ||||
| Lymphoma | Poor [144] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aulas, A.; Finetti, P.; Lyons, S.M.; Bertucci, F.; Birnbaum, D.; Acquaviva, C.; Mamessier, E. Revisiting the Concept of Stress in the Prognosis of Solid Tumors: A Role for Stress Granules Proteins? Cancers 2020, 12, 2470. https://doi.org/10.3390/cancers12092470
Aulas A, Finetti P, Lyons SM, Bertucci F, Birnbaum D, Acquaviva C, Mamessier E. Revisiting the Concept of Stress in the Prognosis of Solid Tumors: A Role for Stress Granules Proteins? Cancers. 2020; 12(9):2470. https://doi.org/10.3390/cancers12092470
Chicago/Turabian StyleAulas, Anaïs, Pascal Finetti, Shawn M. Lyons, François Bertucci, Daniel Birnbaum, Claire Acquaviva, and Emilie Mamessier. 2020. "Revisiting the Concept of Stress in the Prognosis of Solid Tumors: A Role for Stress Granules Proteins?" Cancers 12, no. 9: 2470. https://doi.org/10.3390/cancers12092470