mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme

 , ,
, , 

Abstract
:Simple Summary
Abstract
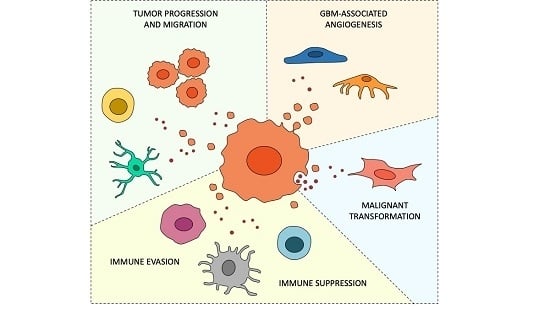
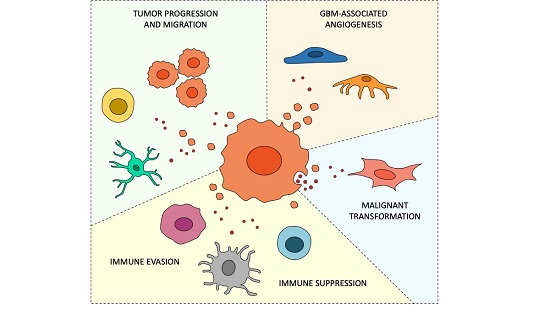{kind=link}
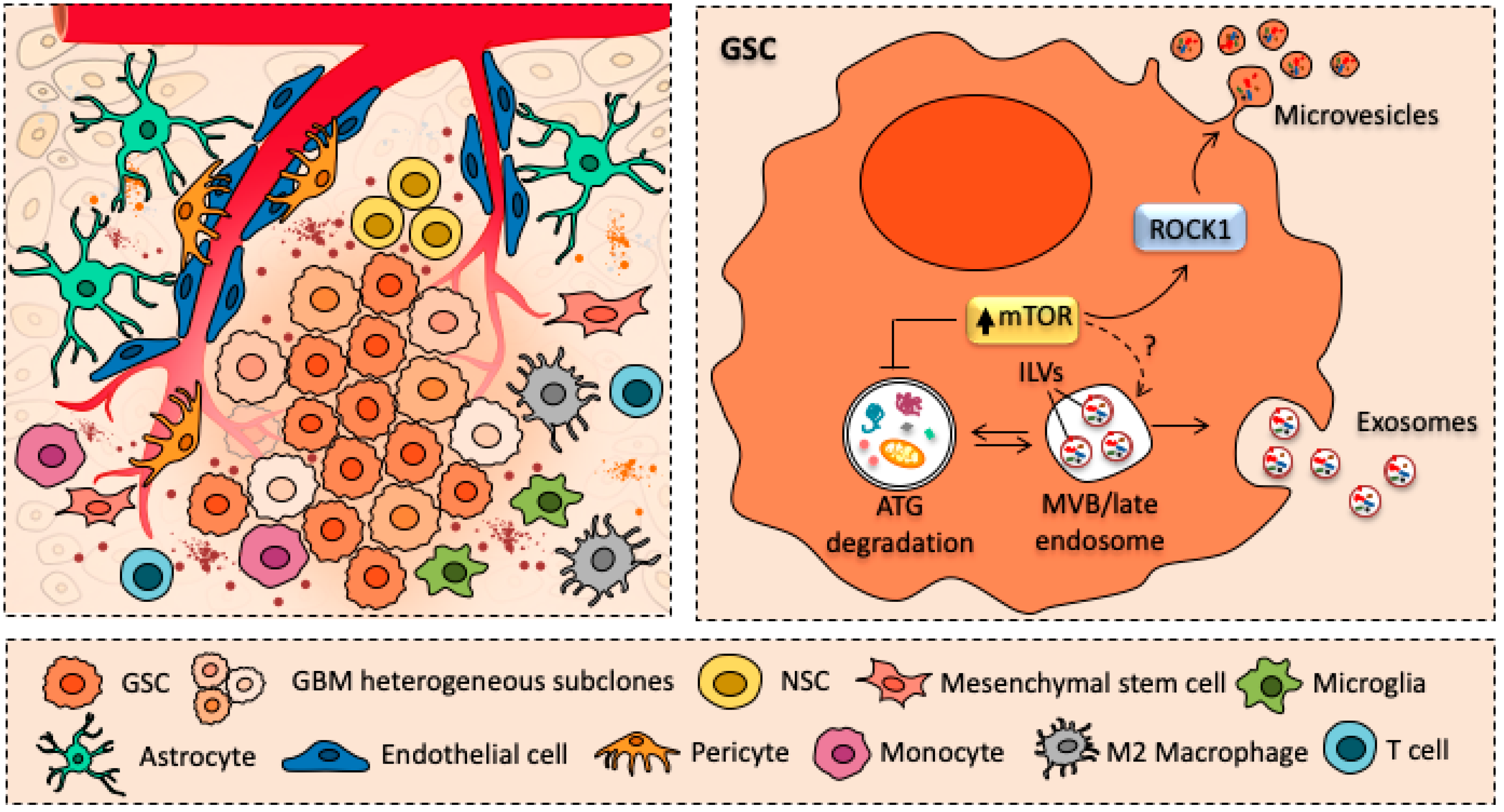{kind=link}
{kind=link}
1. Introduction
2. EV-Mediated Cell-to-Cell Communication in CNS: From Physiological to Pathological Conditions
3. The Role of mTOR in Exosome-Based Cell-to-Cell Communication
4. The Role of mTOR-Dependent GSCs-Derived EVs
5. mTOR-Dependent GSC-Derived EVs Promote GBM Angiogenesis
6. mTOR-Dependent EVs Effects on Glioma-Associated Parenchymal Cells
6.1. The Role of Astrocytes
6.2. The role of Perivascular Pericytes
6.3. The Role of Mesenchymal Stem Cells (MSCs)
7. The Emerging Role of mTOR-Dependent EVs in Immune Escape
8. The Autocrine Role of mTOR-Dependent EVs on GBM Stem Cell Niche
9. Conclusions
Outstanding Questions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| ECs | Endothelial cells |
| EGFR | Epidermal growth factor receptor |
| EPHA2 | Ephrin type A receptor 2 |
| EVs | Extracellular vesicles |
| GA-MSCs | Glioma-associated (GA) mesenchymal stem cells (MSCs) |
| GBM | Glioblastoma multiforme |
| GDEs | GSC-derived exosomes |
| GSCs | Glioma stem-like cells |
| HB-EGF | Heparin-binding EGF-like growth factor |
| hBMSCs | human bone marrow MSCs |
| HBMVECs | human brain microvascular endothelial cells |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| MCP3 | Monocyte-chemotactic protein 3 |
| miRNAs | microRNAs |
| MMPs | Matrix metalloproteinases |
| MSCs | Mesenchymal stem cells |
| MVs | Microvesicles |
| MVBs | Multivesicular bodies |
| NSCs | Neural stem cells |
| NRP1 | Neuropilin |
| ncRNAs | noncoding RNAs |
| PD-1 | Programmed cell death-1 |
| PD-L1 | Programmed death-ligand 1 |
| plxA1 | PlexinA1 |
| rGBM | Recurrent GBM |
| Sema | Semaphorins |
| Sema3A | Semaphorin3A |
| Sema3C | Semapahorin3C |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TF | Tissue factor |
| TIMP-1 | Tissue inhibitor of metalloproteinase-1 |
| TIMP-2 | Tissue inhibitor of metalloproteinase-2 |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Agnihotri, S.; Burrell, K.E.; Wolf, A.; Jalali, S.; Hawkins, C.; Rutka, J.T.; Zadeh, G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Arch. Immunol. Ther. Exp. 2013, 61, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, S.; Yan, J.L.; Torheim, T.; Boonzaier, N.R.; Sinha, R.; Matys, T.; Markowetz, F.; Price, S.J. Characterizing tumor invasiveness of glioblastoma using multiparametric magnetic resonance imaging. J. Neurosurg. 2019, 1–8. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Yan, K.; Yang, K.; Rich, J.N. The evolving landscape of glioblastoma stem cells. Curr. Opin. Neurol. 2013, 26, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Basak, O.; Taylor, V. Stem cells of the adult mammalian brain and their niche. Cell Mol. Life Sci. 2009, 66, 1057–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcantara Llaguno, S.R.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, P.; Martinez Gala, J.; Delmas, C. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Ito, M.; Katsushima, K. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013, 73, 4559–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallik, S.; Benson, R.; Hakim, A.; Rath, G.K. Management of glioblastoma after recurrence: A changing paradigm. J. Egypt Natl. Cancer Inst. 2016, 28, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, E.; Reynolds, B.A.; Vescovi, A.L. Glioma stem cells: Turpis omen in nomen? (The evil in the name?). J. Intern. Med. 2014, 276, 25–40. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar]
- Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Limanaqi, F.; Familiari, P.; Frati, A.; Fornai, F. Prion Protein in Glioblastoma Multiforme. Int. J. Mol. Sci 2019, 20, 5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer. Inst. 2007, 99, 1583–1593. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Zappulli, V.; Friis, K.P.; Fitzpatrick, Z.; Maguire, C.A.; Breakefield, X.O. Extracellular vesicles and intercellular communication within the nervous system. J. Clin. Investig. 2016, 126, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and MYC signaling pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [Green Version]
- Svensson, K.J.; Kucharzewska, P.; Christianson, H.C.; Skold, S.; Lofstedt, T.; Johansson, M.C.; Morgelin, M.; Bengzon, J.; Ruf, W.; Belting, M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Li, C.C.Y.; Eaton, S.A.; Young, P.E.; Lee, M.; Shuttleworth, R.; Humphreys, D.T.; Grau, G.E.; Combes, V.; Bebawy, M.; Gong, J.; et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013, 10, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Hellwinkel, J.E.; Redzic, J.S.; Harland, T.A.; Gunaydin, D.; Anchordoquy, T.J.; Graner, M.W. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 2016, 18, 497–506. [Google Scholar] [CrossRef] [PubMed]
- de Vrij, J.; Maas, S.L.; Kwappenberg, K.M.; Schnoor, R.; Kleijn, A.; Dekker, L.; Luider, T.M.; de Witte, L.D.; Litjens, M.; van Strien, M.E.; et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 2015, 137, 1630–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Bukoreshtliev, N.V.; Barroso, J.F. Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 2007, 581, 2194–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borràs, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [Green Version]
- Holme, P.A.; Solum, N.O.; Brosstad, F.; Røger, M.; Abdelnoor, M. Demonstration of platelet-derived microvesicles in blood from patients with activated coagulation and fibrinolysis using a filtration technique and western blotting. Thromb. Haemost. 1994, 72, 666–671. [Google Scholar]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes-vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteomics 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Yuyama, K.; Igarashi, Y. Physiological and pathological roles of exosomes in the nervous system. Biomol. Concepts 2016, 7, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Krämer-Albers, E.M. Emerging roles of exosomes in neuron-glia communication. Front Physiol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zheng, T.; Zhang, B. Exosomes in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Epple, L.M.; Griffiths, S.G.; Dechkovskaia, A.M.; Dusto, N.L.; White, J.; Ouellette, R.J.; Anchordoquy, T.J.; Bemis, L.T.; Graner, M.W. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS ONE 2012, 7, e42064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haug, B.H.; Hald, Ø.H.; Utnes, P.; Roth, S.A.; Løkke, C.; Flægstad, T.; Einvik, C. Exosome-like Extracellular Vesicles from MYCN-amplified Neuroblastoma Cells Contain Oncogenic miRNAs. Anticancer Res. 2015, 35, 2521–2530. [Google Scholar]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell. 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Ferrucci, M.; Pasquali, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. Alpha-synuclein and autophagy as common steps in neurodegeneration. Parkinsonism Relat. Disord. 2008, 14, S180–S184. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [Green Version]
- Bernard, A.; Klionsky, D.J. Defining the membrane precursor supporting the nucleation of the phagophore. Autophagy 2014, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1 ATG13 FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [Green Version]
- Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses 2017, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.V.; Teixeira, B.L.; Rodrigues, B.R.; Sinigaglia-Coimbra, R.; Porto-Carreiro, I.; Roffé, M.; Hajj, G.N.; Martins, V.R. PRNP/prion protein regulates the secretion of exosomes modulating CAV1/caveolin-1-suppressed autophagy. Autophagy 2016, 12, 2113–2128. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, M.; Seki, T.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Reciprocal Regulation of Chaperone-Mediated Autophagy/Microautophagy and Exosome Release. Biol. Pharm. Bull. 2019, 42, 1394–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, C.M.; Sánchez, D.; Furlán, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro Oncol. 2010, 12, 1205–1219. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef]
- Arcella, F.; Biagioni, M.; Oliva, A.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef]
- Mendiburu-Eliçabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Mammalian target of rapamycin inhibitors, temsirolimus and torin 1, attenuate stemness-associated properties and expression of mesenchymal markers promoted by phorbol-myristate-acetate and oncostatin-M in glioblastoma cells. Tumour. Biol. 2017, 39, 1010428317695921. [Google Scholar] [CrossRef] [Green Version]
- Poehler, A.-M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wei, B.; de Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J. Hepatol. 2020, 30290–30297. [Google Scholar] [CrossRef]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedgwick, A.E.; Clancy, J.W.; Olivia Balmert, M.; D’Souza-Schorey, C. Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci. Rep. 2015, 5, 14748. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Z.; Mashimo, T.; Shen, B.; Nyagilo, J.; Wang, H.; Wang, Y.; Liu, Z.; Mulgaonkar, A.; Hu, X.L.; et al. Gliomas Interact with Non-glioma Brain Cells via Extracellular Vesicles. Cell Rep. 2020, 30, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Liu, S.; Sun, J.; Lan, Q. Glioblastoma microvesicles promote endothelial cell proliferation through Akt/beta-catenin pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 4857–4866. [Google Scholar]
- Cai, Q.; Zhu, A.; Gong, L. Exosomes of glioma cells deliver miR-148a to promote proliferation and metastasis of glioblastoma via targeting CADM1. Bull. Cancer 2018, 105, 643–651. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Y.; Chilukuri, K.; Brady, O.A.; Boulos, M.I.; Kappes, J.C.; Galileo, D.S. L1 stimulation of human glioma cell motility correlates with FAK activation. J. Neurooncol. 2011, 105, 27–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 35, 2615–2623. [Google Scholar] [CrossRef]
- Monteforte, A.; Lam, B.; Sherman, M.B.; Henderson, K.; Sligar, A.D.; Spencer, A.; Tang, B.; Dunn, A.K.; Baker, A.B. Glioblastoma Exosomes for Therapeutic Angiogenesis in Peripheral Ischemia. Tissue Eng. Part A 2017, 23, 1251–1261. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, H.; Xiong, C.; Liu, Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem. Biophys. Res. Commun. 2018, 502, 324–331. [Google Scholar] [CrossRef]
- Le Guelte, A.; Galan-Moya, E.M.; Dwyer, J.; Treps, L.; Kettler, G.; Hebda, J.K.; Dubois, S.; Auffray, C.; Chneiweiss, H.; Bidere, N.; et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J. Cell Sci. 2012, 125, 4137–4146. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Zhao, P.; Li, W.; Shi, Z.M.; Wang, L.; Xu, Q.; Wang, M.; Liu, N.; Liu, L.Z.; Jiang, B.H. MicroRNA-26a promotes tumor growth and angiogenesis in glioma by directly targeting prohibitin. CNS Neurosci. Ther. 2013, 19, 804–812. [Google Scholar] [CrossRef]
- Li, J.; An, G.; Zhang, M.; Ma, Q. Long non-coding RNA TUG1 acts as a miR-26a sponge in human glioma cells. Biochem. Biophys. Res. Commun. 2016, 477, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from glioma-associated mesenchymal stem cells increase the Tumorigenicity of glioma stem-like cells via transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar] [PubMed]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhang, Y.; Cheng, S.; Wu, Z.; Liu, F.; Zhang, J. CD133 positive U87 glioblastoma cells-derived exosomal microRNAs in hypoxia- versus normoxia-microenviroment. J. Neurooncol. 2017, 135, 37–46. [Google Scholar] [CrossRef]
- Pinet, S.; Bessette, B.; Vedrenne, N.; Lacroix, A.; Richard, L.; Jauberteau, M.O.; Battu, S.; Lalloué, F. TrkB-containing exosomes promote the transfer of glioblastoma aggressiveness to YKL-40-inactivated glioblastoma cells. Oncotarget 2016, 7, 50349–50364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef] [Green Version]
- Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers. Int. J. Mol. Sci. 2020, 21, 685. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Tian, Y.; Hu, Y.; Qi, Y.; Tian, N.; Li, S.; Hu, P.; Wu, F.; Wei, Q.; Wei, Z.; et al. Correction: Glioma-associated human endothelial cell-derived extracellular vesicles specifically promote the tumourigenicity of glioma stem cells via CD9. Oncogene 2019, 38, 6958. [Google Scholar] [CrossRef] [Green Version]
- Podergajs, N.; Motaln, H.; Rajčević, U.; Verbovšek, U.; Koršič, M.; Obad, N.; Espedal, H.; Vittori, M.; Herold-Mende, C.; Miletic, H.; et al. Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 2016, 7, 593–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhou, W.; Cheng, L.; Chen, C.; Huang, Z.; Fang, X.; Wu, Q.; He, Z.; Xu, S.; Lathia, J.D.; et al. Tetraspanin CD9 stabilizes gp130 by preventing its ubiquitin-dependent lysosomal degradation to promote STAT3 activation in glioma stem cells. Cell Death Differ. 2017, 24, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes Enhance the Invasion Potential of Glioblastoma Stem-Like Cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603. [Google Scholar] [CrossRef] [Green Version]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE. 2014, 9, e101402. [Google Scholar] [CrossRef] [Green Version]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef] [Green Version]
- Sena, I.F.G.; Paiva, A.E.; Prazeres, P.H.D.M.; Azevedo, P.O.; Lousado, L.; Bhutia, S.K.; Salmina, A.B.; Mintz, A.; Birbrair, A. Glioblastoma-activated pericytes support tumor growth via immunosuppression. Cancer Med. 2018, 7, 1232–1239. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated from Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orbay, H.; Tobita, M.; Mizuno, H. Mesenchymal stem cells isolated from adipose and other tissues: Basic biologicalproperties and clinical applications. Stem Cells Int. 2012, 2012, 461718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavon, L.F.; Sibov, T.T.; de Souza, A.V.; da Cruz, E.F.; Malheiros, S.; Cabral, F.R.; de Souza, J.G.; Boufleur, P.; de Oliveira, D.M.; de Toledo, S.; et al. Tropism of mesenchymal stem cell toward CD133+ stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem Cell Res. Ther. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Luciano, R.; Saracino, R.; Battafarano, G.; Rizzo, C.; Pascucci, L.; Alessandri, G.; Pessina, A.; Perrotta, A.; Fierabracci, A.; et al. Differential effects of extracellular vesicles secreted by mesenchymal stem cells from different sources on glioblastoma cells. Expert Opin. Biol. Ther. 2015, 15, 495–504. [Google Scholar] [CrossRef]
- Lee, H.K.; Finniss, S.; Cazacu, S.; Bucris, E.; Ziv-Av, A.; Xiang, C.; Bobbitt, K.; Rempel, S.A.; Hasselbach, L.; Mikkelsen, T.; et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget 2013, 4, 346–361. [Google Scholar] [CrossRef] [Green Version]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhao, G.; Zhang, Y.; Jiang, H.; Wang, W.; Zhao, D.; Hong, J.; Yu, H.; Qi, L. Mesenchymal stem cell-derived exosomal microRNA-133b suppresses glioma progression via Wnt/β-catenin signaling pathway by targeting EZH2. Stem Cell Res. Ther. 2019, 10, 381. [Google Scholar] [CrossRef] [Green Version]
- Schichor, C.; Birnbaum, T.; Etminan, N.; Schnell, O.; Grau, S.; Miebach, S.; Aboody, K.; Padovan, C.; Straube, A.; Tonn, J.C.; et al. Vascular endothelial growth factor a contributes to glioma-induced migration of human marrow stromal cells (hMSC). Exp. Neurol. 2006, 199, 301–310. [Google Scholar] [CrossRef]
- Xu, F.; Shi, J.; Yu, B.; Ni, W.; Wu, X.; Gu, Z. Chemokines mediate mesenchymal stem cell migration toward gliomas in vitro. Oncol. Rep. 2010, 23, 1561–1567. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Cui, X.; Lu, L.; Chen, G.; Yang, Y.; Hu, Y.; Lu, Y.; Cao, Z.; Wang, Y.; Wang, X. Exosomes from glioma cells induce a tumor-like phenotype in mesenchymal stem cells by activating glycolysis. Stem Cell Res. Ther. 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016, 18, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourkoula, E.; Mangoni, D.; Ius, T.; Pucer, A.; Isola, M.; Musiello, D.; Marzinotto, S.; Toffoletto, B.; Sorrentino, M.; Palma, A.; et al. Glioma-associated stem cells: A novel class of tumor-supporting cells able to predict prognosis of human low-grade gliomas. Stem Cells 2014, 32, 1239–1253. [Google Scholar] [CrossRef]
- Hamerlik, P.; Lathia, J.D.; Rasmussen, R.; Wu, Q.; Bartkova, J.; Lee, M.; Moudry, P.; Bartek, J., Jr.; Fischer, W.; Lukas, J.; et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 2012, 209, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, X.; Zhu, J. VEGF promotes proliferation of human glioblastoma multiforme stem-like cells through VEGF receptor 2. Sci. World J. 2013, 2013, 417413. [Google Scholar] [CrossRef] [Green Version]
- Pace, K.R.; Dutt, R.; Galileo, D.S. Exosomal L1CAM Stimulates Glioblastoma Cell Motility, Proliferation, and Invasiveness. Int. J. Mol. Sci. 2019, 20, 3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; Cuartas, I.; Raventós, C.; Martínez-Ricarte, F.; et al. TGF-beta receptor inhibitors target the CD44(high)/ Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podergajs, N.; Brekka, N.; Radlwimmer, B.; Herold-Mende, C.; Talasila, K.M.; Tiemann, K.; Rajcevic, U.; Lah, T.T.; Bjerkvig, R.; Miletic, H. Expansive growth of two glioblastoma stem-like cell lines is mediated by bFGF and not by EGF. Radiol. Oncol. 2013, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Shoemake, J.; Zhou, W.; Fang, X.; Wu, Q.; Rizzo, A.; Prayson, R.; Bao, S.; Rich, J.N.; Yu, J.S. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Wang, L.; Zhou, Y.; Dong, L.; Ma, W.; Lv, L.; Zhang, J.; Wang, X. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell Mol. Neurobiol. 2020, 40, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Ruaro, M.E.; Sgarra, R.; Bartolini, A.; Caponnetto, F.; Ius, T.; Skrap, M.; Di Loreto, C.; Beltrami, A.P.; Manfioletti, G.; et al. Semaphorin-7A on Exosomes: A Promigratory Signal in the Glioma Microenvironment. Cancers 2019, 11, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagci, T.; Wu, J.K.; Pfannl, R.; Ilag, L.L.; Jay, D.G. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 2009, 28, 3537–3550. [Google Scholar] [CrossRef] [Green Version]
- Frankel, P.; Pellet-Many, C.; Lehtolainen, P.; D’Abaco, G.M.; Tickner, M.L.; Cheng, L.; Zachary, I.C. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. EMBO Rep. 2008, 9, 983–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, I.M.; Yamaji, M.; Britton, G.; Pellet-Many, C.; Lockie, C.; Zachary, I.C.; Frankel, P. Neuropilin-1 signaling through p130Cas tyrosine phosphorylation is essential for growth factor-dependent migration of glioma and endothelial cells. Mol. Cell Biol. 2011, 31, 1174–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, J.; Sun, G.; Meng, H.; Wang, J.; Guan, Y.; Yin, Y.; Zhao, Z.; Dong, X.; Yin, S.; et al. Glioblastoma extracellular vesicles induce the tumour-promoting transformation of neural stem cells. Cancer Lett. 2019, 466, 1–12. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers 2020, 12, 2486. https://doi.org/10.3390/cancers12092486
Ryskalin L, Biagioni F, Lenzi P, Frati A, Fornai F. mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers. 2020; 12(9):2486. https://doi.org/10.3390/cancers12092486
Chicago/Turabian StyleRyskalin, Larisa, Francesca Biagioni, Paola Lenzi, Alessandro Frati, and Francesco Fornai. 2020. "mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme" Cancers 12, no. 9: 2486. https://doi.org/10.3390/cancers12092486
APA StyleRyskalin, L., Biagioni, F., Lenzi, P., Frati, A., & Fornai, F. (2020). mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers, 12(9), 2486. https://doi.org/10.3390/cancers12092486