Insights into Differentiation of Melanocytes from Human Stem Cells and Their Relevance for Melanoma Treatment


Abstract
:Simple Summary
Abstract

1. Introduction
2. Evidence for a Role of Development-Associated Factors during Melanoma Progression
3. In Vivo Differentiation of Melanocytes
4. In Vitro 2D and 3D Differentiation Models for Melanocytes
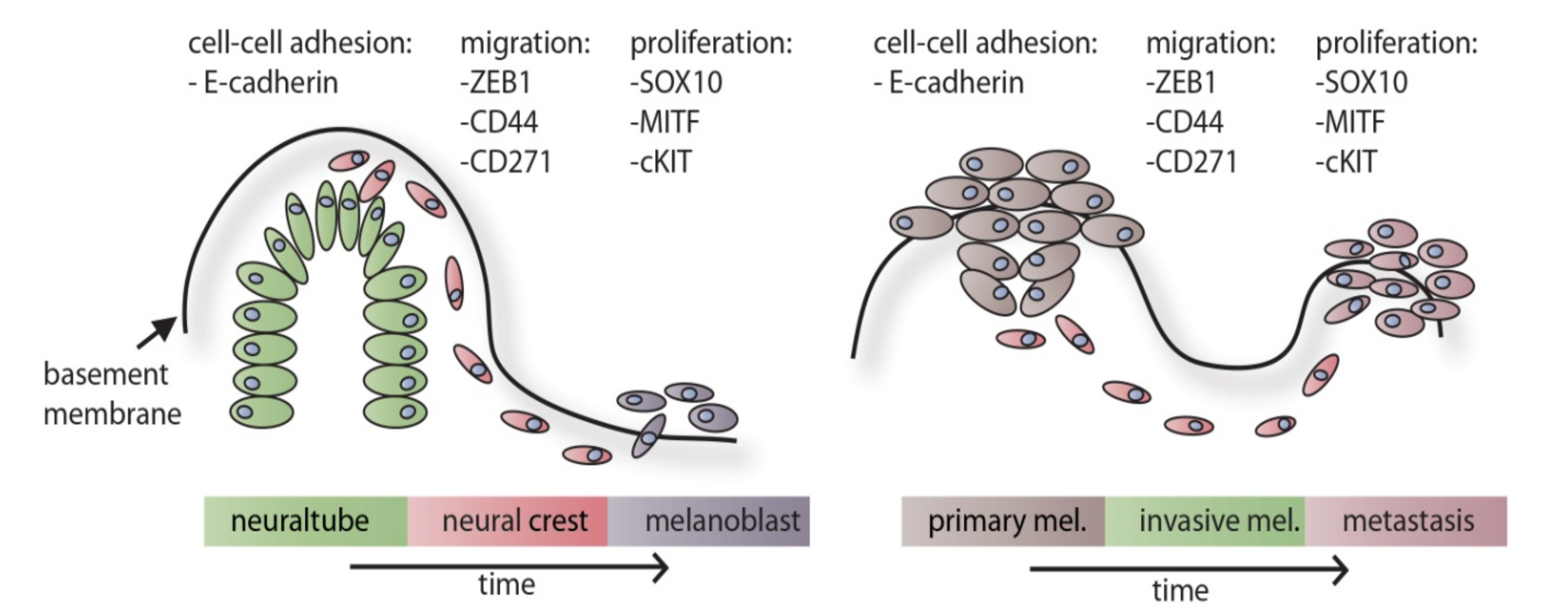
5. Parallels between Neural Crest Development and Melanoma Progression
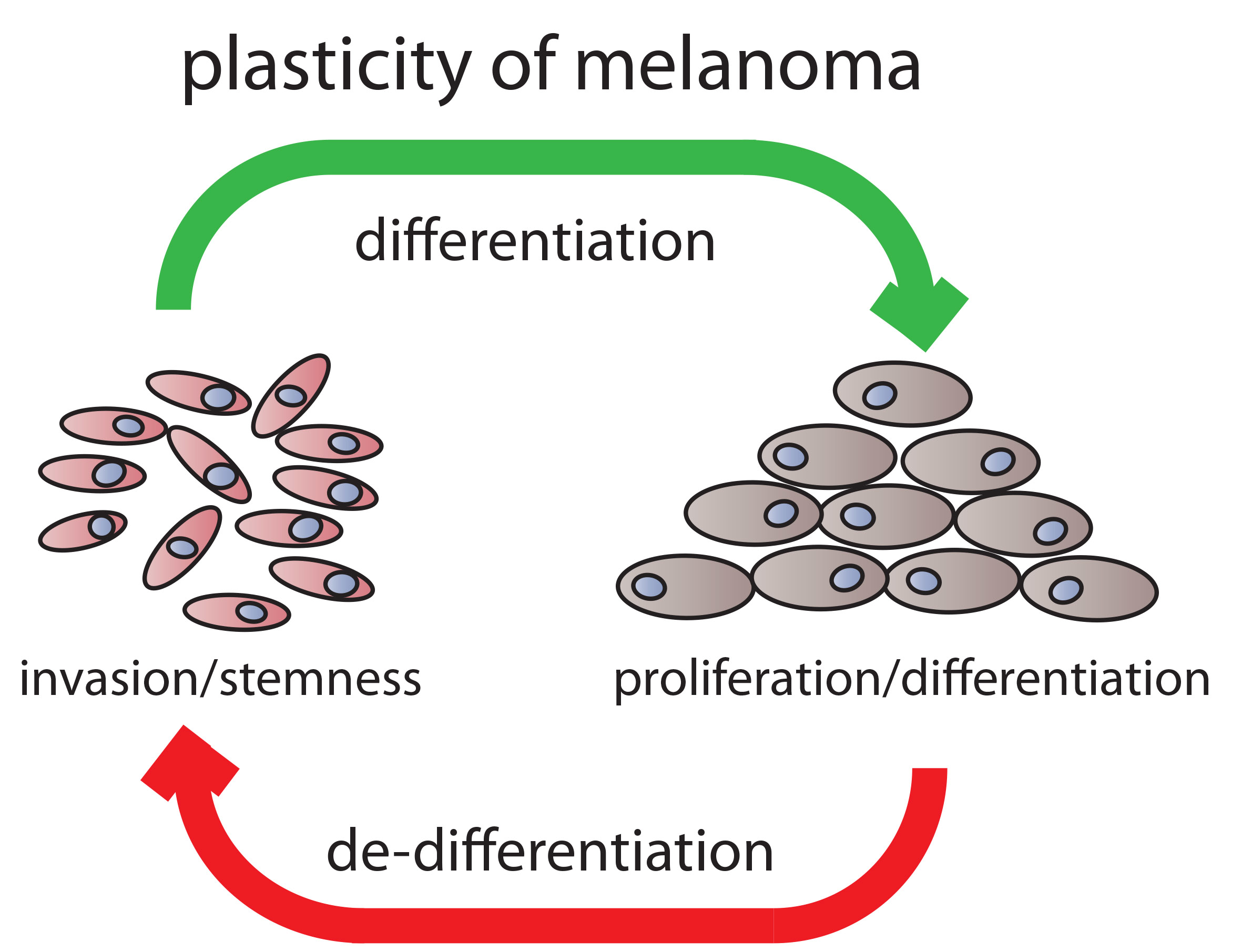
6. A Working Model for Melanoma Progression
7. Conclusion and Possible Therapeutic Options for Future Melanoma Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lübbe, J.; Reichel, M.; Burg, G.; Kleihues, P. Absence of p53 gene mutations in cutaneous melanoma. J. Invest. Dermatol. 1994, 102, 819–821. [Google Scholar] [CrossRef] [Green Version]
- Piepkorn, M. Melanoma genetics: An update with focus on the CDKN2A(p16)/ARF tumor suppressors. J. Am. Acad. Dermatol. 2000, 42, 705–722, quiz 723. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [PubMed]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Steder, M.; Alla, V.; Meier, C.; Spitschak, A.; Pahnke, J.; Fürst, K.; Kowtharapu, B.S.; Engelmann, D.; Petigk, J.; Egberts, F.; et al. DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell 2013, 24, 512–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. (Basel) 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Kerosuo, L.; Bronner-Fraser, M. What is bad in cancer is good in the embryo: Importance of EMT in neural crest development. Semin. Cell Dev. Biol. 2012, 23, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Thompson, E.W.; Haviv, I. The social aspects of EMT-MET plasticity. Nat. Med. 2011, 17, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [Green Version]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.-R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C. Disassembling adherens junctions: Breaking up is hard to do. Trends Cell Biol. 2005, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Stow, J.L. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004, 14, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.M.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Grände, M.; Franzen, A.; Karlsson, J.-O.; Ericson, L.E.; Heldin, N.-E.; Nilsson, M. Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 2002, 115, 4227–4236. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Comijn, J.; De Craene, B.; Vermassen, P.; Bruyneel, E.; Andersen, H.; Tulchinsky, E.; Van Roy, F.; Berx, G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005, 33, 6566–6578. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Chen, H.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumour Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerschenson, M.; Graves, K.; Carson, S.D.; Wells, R.S.; Pierce, G.B. Regulation of melanoma by the embryonic skin. Proc. Natl. Acad. Sci. USA 1986, 83, 7307–7310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulesa, P.M.; Kasemeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.B.; Hendrix, M.J.C. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 3752–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.S.; Heilmann, S.; Kansler, E.R.; Zhang, Y.; Zimmer, M.; Ratnakumar, K.; Bowman, R.L.; Simon-Vermot, T.; Fennell, M.; Garippa, R.; et al. Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat. Commun. 2017, 8, 14343. [Google Scholar] [CrossRef] [Green Version]
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Santagati, F.; Rijli, F.M. Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci. 2003, 4, 806–818. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar]
- Lallier, T.E. Cell lineage and cell migration in the neural crest. Ann. N. Y. Acad. Sci. 1991, 615, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chan, A.K.K.; Sham, M.H.; Burns, A.J.; Chan, W.Y. Analysis of the sacral neural crest cell contribution to the hindgut enteric nervous system in the mouse embryo. Gastroenterology 2011, 141, 992–1002.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.F. The Neural Crest, 6th ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Müller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, S.F.; Fesyuk, A. On the development of neurocutaneous units--implications for the histogenesis of congenital, acquired, and dysplastic nevi. Am. J. Dermatopathol. 2012, 34, 60–81. [Google Scholar] [CrossRef] [PubMed]
- Gleason, B.C.; Crum, C.P.; Murphy, G.F. Expression patterns of MITF during human cutaneous embryogenesis: Evidence for bulge epithelial expression and persistence of dermal melanoblasts. J. Cutan. Pathol. 2008, 35, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Dupin, E.; Real, C.; Glavieux-Pardanaud, C.; Vaigot, P.; Le Douarin, N.M. Reversal of developmental restrictions in neural crest lineages: Transition from Schwann cells to glial-melanocytic precursors in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 5229–5233. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Toyoda, M.; Yaar, M.; Bhawan, J.; Avila, E.M.; Penner, I.R.; Gilchrest, B.A. Innervation of melanocytes in human skin. J. Exp. Med. 1996, 184, 1385–1395. [Google Scholar] [CrossRef]
- Vance, K.W.; Goding, C.R. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res. 2004, 17, 318–325. [Google Scholar] [CrossRef]
- Hirobe, T. How are proliferation and differentiation of melanocytes regulated? Pigment. Cell Melanoma Res. 2011, 24, 462–478. [Google Scholar] [CrossRef]
- Ernfors, P. Cellular origin and developmental mechanisms during the formation of skin melanocytes. Exp. Cell Res. 2010, 316, 1397–1407. [Google Scholar] [CrossRef]
- Harris, M.L.; Erickson, C.A. Lineage specification in neural crest cell pathfinding. Dev. Dyn. 2007, 236, 1–19. [Google Scholar] [CrossRef]
- Dupin, E.; Sommer, L. Neural crest progenitors and stem cells: From early development to adulthood. Dev. Biol. 2012, 366, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Hari, L.; Miescher, I.; Shakhova, O.; Suter, U.; Chin, L.; Taketo, M.; Richardson, W.D.; Kessaris, N.; Sommer, L. Temporal control of neural crest lineage generation by Wnt/β-catenin signaling. Development 2012, 139, 2107–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, C.D.; Raible, D.W. Mechanisms for reaching the differentiated state: Insights from neural crest-derived melanocytes. Semin. Cell Dev. Biol. 2009, 20, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, G.G.; Horstmann, M.; Widlund, H.R.; Du, J.; Motyckova, G.; Nishimura, E.K.; Lin, Y.-L.; Ramaswamy, S.; Avery, W.; Ding, H.-F.; et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 2002, 109, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Sommer, L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 2011, 24, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Montague, P.M.; Hearing, V.J. Anti-T4-tyrosinase monoclonal antibodies--specific markers for pigmented melanocytes. J. Invest. Dermatol. 1985, 85, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larribere, L.; Utikal, J. De- and re-differentiation of the melanocytic lineage. Eur. J. Cell Biol. 2014, 93, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Mica, Y.; Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 2013, 3, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissan, X.; Larribere, L.; Saidani, M.; Hurbain, I.; Delevoye, C.; Feteira, J.; Lemaitre, G.; Peschanski, M.; Baldeschi, C. Functional melanocytes derived from human pluripotent stem cells engraft into pluristratified epidermis. Proc. Natl. Acad. Sci. USA 2011, 108, 14861–14866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.D.; Jayasena, C.S.; Nie, S.; Bronner, M.E. Neural crest specification: Tissues, signals, and transcription factors. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Münst, S.; Koch, P.; Kesavan, J.; Alexander-Mays, M.; Münst, B.; Blaess, S.; Brüstle, O. In vitro segregation and isolation of human pluripotent stem cell-derived neural crest cells. Methods 2018, 133, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Yoshida, S.; Inagaki, E.; Hatou, S.; Tsubota, K.; Takahashi, M.; Shimmura, S.; Sugita, S. Immunological Properties of Neural Crest Cells Derived from Human Induced Pluripotent Stem Cells. Stem Cells Dev. 2019, 28, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Larribere, L.; Wu, H.; Novak, D.; Galach, M.; Bernhardt, M.; Orouji, E.; Weina, K.; Knappe, N.; Sachpekidis, C.; Umansky, L.; et al. NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment Cell Melanoma Res. 2015, 28, 407–416. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lim, H.; Li, Z.; Oh, Y.; Kovlyagina, I.; Choi, I.Y.; Dong, X.; Lee, G. Generation of multipotent induced neural crest by direct reprogramming of human postnatal fibroblasts with a single transcription factor. Cell Stem Cell 2014, 15, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Qi, Y.; Mica, Y.; Lee, G.; Zhang, X.-J.; Niu, L.; Bilsland, J.; Cao, L.; Stevens, E.; Whiting, P.; et al. Combined small-molecule inhibition accelerates developmental timing’ ‘ and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 2012, 30, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Fattahi, F.; Steinbeck, J.A.; Kriks, S.; Tchieu, J.; Zimmer, B.; Kishinevsky, S.; Zeltner, N.; Mica, Y.; El-Nachef, W.; Zhao, H.; et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 2016, 531, 105–109. [Google Scholar] [CrossRef]
- Schlieve, C.R.; Fowler, K.L.; Thornton, M.; Huang, S.; Hajjali, I.; Hou, X.; Grubbs, B.; Spence, J.R.; Grikscheit, T.C. Neural Crest Cell Implantation Restores Enteric Nervous System Function and Alters the Gastrointestinal Transcriptome in Human Tissue-Engineered Small Intestine. Stem Cell Rep. 2017, 9, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Miller, M.L.; McHenry, L.K.; Zheng, T.; Zhen, Q.; Ilkhanizadeh, S.; Conklin, B.R.; Bronner, M.E.; Weiss, W.A. Generating trunk neural crest from human pluripotent stem cells. Sci. Rep. 2016, 6, 19727. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, N.; Berx, G. From neural crest cells to melanocytes: Cellular plasticity during development and beyond. Cell. Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef]
- Pala, L.; Conforti, F.; Cocorocchio, E.; Ferrucci, P.F. Extensive vitiligo associated to response to c-kit inhibitor in metastatic mucosal melanoma. Anticancer Drugs 2020, 31, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.L.; Baxter, L.L.; Loftus, S.K.; Pavan, W.J. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010, 23, 496–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Rabbani, C.C.; Gao, H.; Steinhart, M.R.; Woodruff, B.M.; Pflum, Z.E.; Kim, A.; Heller, S.; Liu, Y.; Shipchandler, T.Z.; et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature 2020, 582, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Kalcheim, C. Association between the cell cycle and neural crest delamination through specific regulation of G1/S transition. Dev. Cell 2002, 3, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Théveneau, E.; Duband, J.-L.; Altabef, M. Ets-1 confers cranial features on neural crest delamination. PLoS ONE 2007, 2, e1142. [Google Scholar] [CrossRef] [PubMed]
- Ridenour, D.A.; McLennan, R.; Teddy, J.M.; Semerad, C.L.; Haug, J.S.; Kulesa, P.M. The neural crest cell cycle is related to phases of migration in the head. Development 2014, 141, 1095–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.M.; Hernandez, S.; Giang, B.; Chabot, C.; Hernandez, J.; de Bellard, M.E. Molecular events controlling cessation of trunk neural crest migration and onset of differentiation. Front. Cell Dev. Biol. 2020, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, M.L.; Palma, C.d.S.; Thomé, C.H.; Lanfredi, G.P.; Poersch, A.; Faça, V.M. Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control. J. Proteom. 2017, 151, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ye, F.; Li, Q.; Tamiya, S.; Darling, D.S.; Kaplan, H.J.; Dean, D.C. Zeb1 represses Mitf and regulates pigment synthesis, cell proliferation, and epithelial morphology. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5080–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, E.; Yokoyama, S.; Levy, C.; Khaled, M.; Igras, V.; Lin, R.J.; Lee, S.; Widlund, H.R.; Granter, S.R.; Kung, A.L.; et al. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc. Natl. Acad. Sci. USA 2011, 108, E924–E933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goding, C.R. Mitf from neural crest to melanoma: Signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000, 14, 1712–1728. [Google Scholar] [PubMed]
- Shakhova, O.; Zingg, D.; Schaefer, S.M.; Hari, L.; Civenni, G.; Blunschi, J.; Claudinot, S.; Okoniewski, M.; Beermann, F.; Mihic-Probst, D.; et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat. Cell Biol. 2012, 14, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eccles, M.R.; He, S.; Ahn, A.; Slobbe, L.J.; Jeffs, A.R.; Yoon, H.-S.; Baguley, B.C. MITF and PAX3 play distinct roles in melanoma cell migration; outline of a “genetic switch” theory involving MITF and PAX3 in proliferative and invasive phenotypes of melanoma. Front. Oncol. 2013, 3, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, J.; Pan, L.; Li, H.; Rao, C.; Zhang, X.; Niu, G.; Qu, J.; Hou, L. BMP4 is required for the initial expression of MITF in melanocyte precursor differentiation from embryonic stem cells. Exp. Cell Res. 2014, 320, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.M.; Morrison, J.A.; Kulesa, P.M. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res. 2012, 25, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herraiz, C.; Jiménez-Cervantes, C.; Sánchez-Laorden, B.; García-Borrón, J.C. Functional interplay between secreted ligands and receptors in melanoma. Semin. Cell Dev. Biol. 2018, 78, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Piras, F.; Perra, M.T.; Murtas, D.; Minerba, L.; Floris, C.; Maxia, C.; Demurtas, P.; Ugalde, J.; Ribatti, D.; Sirigu, P. The stem cell marker nestin predicts poor prognosis in human melanoma. Oncol. Rep. 2009, 23, 17–24. [Google Scholar] [CrossRef]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Fodstad, O.; Lorico, A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 2008, 26, 3008–3017. [Google Scholar] [CrossRef] [Green Version]
- Li, Z. CD133: A stem cell biomarker and beyond. Exp. Hematol. Oncol. 2013, 2, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipp, F.V.; Li, C.; Boiko, A.D. CD271 is a molecular switch with divergent roles in melanoma and melanocyte development. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmer, T.; Welte, Y.; Behrens, D.; Fichtner, I.; Przybilla, D.; Wruck, W.; Yaspo, M.-L.; Lehrach, H.; Schäfer, R.; Regenbrecht, C.R.A. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS ONE 2014, 9, e92596. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Graham, V.; Khudyakov, J.; Ellis, P.; Pevny, L. SOX2 functions to maintain neural progenitor identity. Neuron 2003, 39, 749–765. [Google Scholar] [CrossRef] [Green Version]
- Girouard, S.D.; Laga, A.C.; Mihm, M.C.; Scolyer, R.A.; Thompson, J.F.; Zhan, Q.; Widlund, H.R.; Lee, C.-W.; Murphy, G.F. SOX2 contributes to melanoma cell invasion. Lab. Invest. 2012, 92, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Laga, A.C.; Zhan, Q.; Weishaupt, C.; Ma, J.; Frank, M.H.; Murphy, G.F. SOX2 and nestin expression in human melanoma: An immunohistochemical and experimental study. Exp. Dermatol. 2011, 20, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Hüser, L.; Sachindra, S.; Granados, K.; Federico, A.; Larribère, L.; Novak, D.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int. J. Cancer 2018, 143, 3131–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosanò, L.; Spinella, F.; Genovesi, G.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-B receptor blockade inhibits molecular effectors of melanoma cell progression. J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. S1), S136–S139. [Google Scholar] [CrossRef]
- Wu, R.-L.; Sedlmeier, G.; Kyjacova, L.; Schmaus, A.; Philipp, J.; Thiele, W.; Garvalov, B.K.; Sleeman, J.P. Hyaluronic acid-CD44 interactions promote BMP4/7-dependent Id1/3 expression in melanoma cells. Sci. Rep. 2018, 8, 14913. [Google Scholar] [CrossRef] [Green Version]
- Bedogni, B.; Welford, S.M.; Cassarino, D.S.; Nickoloff, B.J.; Giaccia, A.J.; Powell, M.B. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell 2005, 8, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Liu, Y.; Li, Y.; Peng, J.; Li, L.; Liu, J.; Shen, X.; Shen, G.; Tu, Y. Hypoxia: Dual effect on the expression of transferrin receptor in human melanoma A375 cell line. Exp. Dermatol. 2007, 16, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, J.; Li, Y.; Yang, J.; Yu, Y.; Wang, M.; Xu, X.; Huang, C.; Huang, W.; Dong, J.; et al. Targeting hypoxia-inducible factor-1alpha with Tf-PEI-shRNA complex via transferrin receptor-mediated endocytosis inhibits melanoma growth. Mol. Ther. 2009, 17, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Tudrej, K.B.; Czepielewska, E.; Kozłowska-Wojciechowska, M. SOX10-MITF pathway activity in melanoma cells. Arch. Med. Sci. 2017, 13, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [Green Version]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef] [Green Version]
- Cronin, J.C.; Wunderlich, J.; Loftus, S.K.; Prickett, T.D.; Wei, X.; Ridd, K.; Vemula, S.; Burrell, A.S.; Agrawal, N.S.; Lin, J.C.; et al. Frequent mutations in the MITF pathway in melanoma. Pigment Cell Melanoma Res. 2009, 22, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Eves, P.; Haycock, J.; Layton, C.; Wagner, M.; Kemp, H.; Szabo, M.; Morandini, R.; Ghanem, G.; García-Borrón, J.C.; Jiménez-Cervantes, C.; et al. Anti-inflammatory and anti-invasive effects of alpha-melanocyte-stimulating hormone in human melanoma cells. Br. J. Cancer 2003, 89, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, T.; Assmann, V.; Fieber, C.; Termeer, C.; Herrlich, P.; Hofmann, M.; Simon, J.C. CD44 is the principal mediator of hyaluronic-acid-induced melanoma cell proliferation. J. Invest. Dermatol. 2001, 116, 93–101. [Google Scholar] [PubMed]
- Ahrens, T.; Sleeman, J.P.; Schempp, C.M.; Howells, N.; Hofmann, M.; Ponta, H.; Herrlich, P.; Simon, J.C. Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid. Oncogene 2001, 20, 3399–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.; Byers, H.R.; Vink, J.; Stamenkovic, I. CD44H regulates tumor cell migration on hyaluronate-coated substrate. J. Cell Biol. 1992, 118, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Karjalainen, J.M.; Tammi, R.H.; Tammi, M.I.; Eskelinen, M.J.; Agren, U.M.; Parkkinen, J.J.; Alhava, E.M.; Kosma, V.M. Reduced level of CD44 and hyaluronan associated with unfavorable prognosis in clinical stage I cutaneous melanoma. Am. J. Pathol. 2000, 157, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [Green Version]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Jaksch, M.; Múnera, J.; Bajpai, R.; Terskikh, A.; Oshima, R.G. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 2008, 68, 7882–7886. [Google Scholar] [CrossRef] [Green Version]
- Le, N.T.V.; Richardson, D.R. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim. Biophys. Acta 2002, 1603, 31–46. [Google Scholar] [CrossRef]
- Kang, J.S.; Cho, D.; Kim, Y.-I.; Hahm, E.; Kim, Y.S.; Jin, S.N.; Kim, H.N.; Kim, D.; Hur, D.; Park, H.; et al. Sodium ascorbate (vitamin C) induces apoptosis in melanoma cells via the down-regulation of transferrin receptor dependent iron uptake. J. Cell. Physiol. 2005, 204, 192–197. [Google Scholar] [CrossRef]
- Buscà, R.; Berra, E.; Gaggioli, C.; Khaled, M.; Bille, K.; Marchetti, B.; Thyss, R.; Fitsialos, G.; Larribère, L.; Bertolotto, C.; et al. Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J. Cell Biol. 2005, 170, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, T.; Braig, S.; Bosserhoff, A.-K. Bone morphogenetic proteins induce expression of metalloproteinases in melanoma cells and fibroblasts. Eur. J. Cancer 2008, 44, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, T.; Poser, I.; Soncin, F.; Bataille, F.; Moser, M.; Bosserhoff, A.-K. Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 2005, 65, 448–456. [Google Scholar] [PubMed]
- Lu, C.; Kerbel, R.S. Interleukin-6 undergoes transition from paracrine growth inhibitor to autocrine stimulator during human melanoma progression. J. Cell Biol. 1993, 120, 1281–1288. [Google Scholar] [CrossRef]
- Moretti, S.; Chiarugi, A.; Semplici, F.; Salvi, A. Serum imbalance of cytokines in melanoma patients. Melanoma Res. 2001, 11, 395–399. [Google Scholar] [CrossRef]
- Colombo, M.P.; Maccalli, C.; Mattei, S.; Melani, C.; Radrizzani, M.; Parmiani, G. Expression of cytokine genes, including IL-6, in human malignant melanoma cell lines. Melanoma Res. 1992, 2, 181–189. [Google Scholar] [CrossRef]
- Kortylewski, M.; Heinrich, P.C.; Mackiewicz, A.; Schniertshauer, U.; Klingmüller, U.; Nakajima, K.; Hirano, T.; Horn, F.; Behrmann, I. Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene 1999, 18, 3742–3753. [Google Scholar] [CrossRef] [Green Version]
- Brychtova, S.; Fiuraskova, M.; Hlobilková, A.; Brychta, T.; Hirnak, J. Nestin expression in cutaneous melanomas and melanocytic nevi. J. Cutan. Pathol. 2007, 34, 370–375. [Google Scholar] [CrossRef]
- Le Coz, V.; Zhu, C.; Devocelle, A.; Vazquez, A.; Boucheix, C.; Azzi, S.; Gallerne, C.; Eid, P.; Lecourt, S.; Giron-Michel, J. IGF-1 contributes to the expansion of melanoma-initiating cells through an epithelial-mesenchymal transition process. Oncotarget 2016, 7, 82511–82527. [Google Scholar] [CrossRef]
- Hilmi, C.; Larribere, L.; Giuliano, S.; Bille, K.; Ortonne, J.-P.; Ballotti, R.; Bertolotto, C. IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin. J. Invest. Dermatol. 2008, 128, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Jamal, S.; Schneider, R.J. UV-induction of keratinocyte endothelin-1 downregulates E-cadherin in melanocytes and melanoma cells. J. Clin. Invest. 2002, 110, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1alpha in ovarian carcinoma cells. J. Biol. Chem. 2002, 277, 27850–27855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Decandia, S.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res. 2007, 67, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahav, R. Endothelin receptor B is required for the expansion of melanocyte precursors and malignant melanoma. Int. J. Dev. Biol. 2005, 49, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Lassam, N.; Bickford, S. Loss of c-kit expression in cultured melanoma cells. Oncogene 1992, 7, 51–56. [Google Scholar]
- Montone, K.T.; van Belle, P.; Elenitsas, R.; Elder, D.E. Proto-oncogene c-kit expression in malignant melanoma: Protein loss with tumor progression. Mod. Pathol. 1997, 10, 939–944. [Google Scholar]
- Huang, S.; Luca, M.; Gutman, M.; McConkey, D.J.; Langley, K.E.; Lyman, S.D.; Bar-Eli, M. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 1996, 13, 2339–2347. [Google Scholar]
- Giehl, K.A.; Nägele, U.; Volkenandt, M.; Berking, C. Protein expression of melanocyte growth factors (bFGF, SCF) and their receptors (FGFR-1, c-kit) in nevi and melanoma. J. Cutan. Pathol. 2007, 34, 7–14. [Google Scholar] [CrossRef]
- Prignano, F.; Gerlini, G.; Salvatori, B.; Orlando, C. Stem cell factor affects tumour progression markers in metastatic melanoma cells. Clin. Exp. Metastasis 2006, 23, 177–186. [Google Scholar] [CrossRef]
- Welker, P.; Schadendorf, D.; Artuc, M.; Grabbe, J.; Henz, B.M. Expression of SCF splice variants in human melanocytes and melanoma cell lines: Potential prognostic implications. Br. J. Cancer 2000, 82, 1453–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Jove, R.; Yu, H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005, 24, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Cheli, Y.; Bonet, C.; Bonazzi, V.F.; Allegra, M.; Giuliano, S.; Bille, K.; Bahadoran, P.; Giacchero, D.; Lacour, J.P.; et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 2013, 4, 2212–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzner, T.; Bedeir, A.; Held, G.; Peter-Vörösmarty, B.; Ghassemi, S.; Heinzle, C.; Spiegl-Kreinecker, S.; Marian, B.; Holzmann, K.; Grasl-Kraupp, B.; et al. Fibroblast growth factor receptors as therapeutic targets in human melanoma: Synergism with BRAF inhibition. J. Invest. Dermatol. 2011, 131, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, M.S.; Faraone, D.; D’Arcangelo, D.; De Marchis, F.; Toietta, G.; Ribatti, D.; Parazzoli, A.; Colombo, P.; Capogrossi, M.C.; Facchiano, A. The FGF-2-derived peptide FREG inhibits melanoma growth in vitro and in vivo. Mol. Ther. 2011, 19, 266–273. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, S.; Li, Q.; Wang, C.; Lai, X.; Chen, X.; Wang, R.; Di, J.; Li, T.; Wang, W.; et al. The FGF2-binding peptide P7 inhibits melanoma growth in vitro and in vivo. J. Cancer Res. Clin. Oncol. 2012, 138, 1321–1328. [Google Scholar] [CrossRef]
- Xiao, L.; Yang, S.; Hao, J.; Yuan, X.; Luo, W.; Jiang, L.; Hu, Y.; Fu, Z.; Zhang, Y.; Zou, C. Endostar attenuates melanoma tumor growth via its interruption of b-FGF mediated angiogenesis. Cancer Lett. 2015, 359, 148–154. [Google Scholar] [CrossRef]
- de Aguiar, R.B.; Parise, C.B.; Souza, C.R.T.; Braggion, C.; Quintilio, W.; Moro, A.M.; Navarro Marques, F.L.; Buchpiguel, C.A.; Chammas, R.; de Moraes, J.Z. Blocking FGF2 with a new specific monoclonal antibody impairs angiogenesis and experimental metastatic melanoma, suggesting a potential role in adjuvant settings. Cancer Lett. 2016, 371, 151–160. [Google Scholar] [CrossRef]
- Belleudi, F.; Cardinali, G.; Kovacs, D.; Picardo, M.; Torrisi, M.R. KGF Promotes Paracrine Activation of the SCF/c-KIT Axis from Human Keratinocytes to Melanoma Cells. Transl. Oncol. 2010, 3, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Albino, A.P.; Davis, B.M.; Nanus, D.M. Induction of growth factor RNA expression in human malignant melanoma: Markers of transformation. Cancer Res. 1991, 51, 4815–4820. [Google Scholar] [PubMed]
- Ceccarelli, S.; Romano, F.; Angeloni, A.; Marchese, C. Potential dual role of KGF/KGFR as a target option in novel therapeutic strategies for the treatment of cancers and mucosal damages. Expert Opin. Ther. Targets 2012, 16, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Straume, O.; Akslen, L.A. Strong expression of ID1 protein is associated with decreased survival, increased expression of ephrin-A1/EPHA2, and reduced thrombospondin-1 in malignant melanoma. Br. J. Cancer 2005, 93, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillo, E.; Diaz-Lopez, A.; Cuevas, E.P.; Moreno-Bueno, G.; Peinado, H.; Montes, A.; Santos, V.; Portillo, F.; Cano, A. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS ONE 2013, 8, e59948. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Herlyn, M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Enk, A.H.; Jonuleit, H.; Saloga, J.; Knop, J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int. J. Cancer 1997, 73, 309–316. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, B.; Yin, C.; Liu, W.; Han, C.; Chen, B.; Liu, T.; Li, X.; Chen, X.; Li, C.; et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Block, M.S.; Nevala, W.K.; Pang, Y.-P.; Allred, J.B.; Strand, C.; Markovic, S.N. A pilot clinical trial testing topical resiquimod and a xenopeptide as immune adjuvants for a melanoma vaccine targeting MART-1. Melanoma Res. 2019, 29, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Miles, S.L.; Fischer, A.P.; Joshi, S.J.; Niles, R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Cancer 2015, 15, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roomi, M.W.; Roomi, N.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Inhibition of pulmonary metastasis of melanoma b16fo cells in C57BL/6 mice by a nutrient mixture consisting of ascorbic Acid, lysine, proline, arginine, and green tea extract. Exp. Lung Res. 2006, 32, 517–530. [Google Scholar] [CrossRef]
- Schlegel, N.C.; von Planta, A.; Widmer, D.S.; Dummer, R.; Christofori, G. PI3K signalling is required for a TGFβ-induced epithelial-mesenchymal-like transition (EMT-like) in human melanoma cells. Exp. Dermatol. 2015, 24, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Delmas, V.; Möller, M.; Sextius, P.; André, J.; Menashi, S.; Larue, L.; Mauviel, A. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene 2005, 24, 7624–7629. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Klotzsche-von Ameln, A.; Muschter, A.; Mamlouk, S.; Kalucka, J.; Prade, I.; Franke, K.; Rezaei, M.; Poitz, D.M.; Breier, G.; Wielockx, B. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFβ. Cancer Res. 2011, 71, 3306–3316. [Google Scholar] [CrossRef] [Green Version]
- Winder, M.; Virós, A. Mechanisms of drug resistance in melanoma. Handb. Exp. Pharmacol. 2018, 249, 91–108. [Google Scholar]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Richard, G.; Dalle, S.; Monet, M.-A.; Ligier, M.; Boespflug, A.; Pommier, R.M.; de la Fouchardière, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef] [PubMed]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the challenge of targeting cancer stem cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and in vitro evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Category | Name | Proliferative, Differentiated | Migrative, Progenitor | References |
|---|---|---|---|---|
| TF | SOX10 | + + | + | [74,98] |
| TF | SOX2 | + | + + | [90,91,99] |
| TF | MITF | + + + | + | [73,100,101] |
| hormone | a-MSH/Melanocortin | + + + | + | [80,102] |
| receptor/ligand | CD44/Hyaluronic acid | + | + + | [94,103,104,105,106] |
| receptor | CD271 | + | + + + | [86,87,107] |
| receptor | CD133 | + | + + | [84,108,109] |
| co-enzyme | Iron | + + | + | [110,111] |
| TF | HIF-1α | + | + + | [96,97,112] |
| ligand | BMP-4 | + + | + + | [78,113,114] |
| cytokine | IL-6 | + | + + + | [115,116,117,118] |
| filament | Nestin | + | + + | [81,119] |
| hormone | IGF-1 | + | + | [120,121] |
| ligand | Endothelin 1, 3 | + | + + | [122,123] |
| receptor | Endothelin receptor | + + | + | [124,125] |
| receptor | c-kit | + + | + | [126,127,128,129] |
| ligand | SCF | + + | + | [130,131,132] |
| TF | STAT3 | + | + + | [133,134,135] |
| ligand | FGF2 | + + + | + | [136,137,138,139,140] |
| ligand | FGF7 | + + | + | [141,142,143] |
| TF | Id1, Id3 | + | + + | [94,144,145] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirea, M.A.; Eckensperger, S.; Hengstschläger, M.; Mikula, M. Insights into Differentiation of Melanocytes from Human Stem Cells and Their Relevance for Melanoma Treatment. Cancers 2020, 12, 2508. https://doi.org/10.3390/cancers12092508
Mirea MA, Eckensperger S, Hengstschläger M, Mikula M. Insights into Differentiation of Melanocytes from Human Stem Cells and Their Relevance for Melanoma Treatment. Cancers. 2020; 12(9):2508. https://doi.org/10.3390/cancers12092508
Chicago/Turabian StyleMirea, Madalina A., Stefan Eckensperger, Markus Hengstschläger, and Mario Mikula. 2020. "Insights into Differentiation of Melanocytes from Human Stem Cells and Their Relevance for Melanoma Treatment" Cancers 12, no. 9: 2508. https://doi.org/10.3390/cancers12092508
APA StyleMirea, M. A., Eckensperger, S., Hengstschläger, M., & Mikula, M. (2020). Insights into Differentiation of Melanocytes from Human Stem Cells and Their Relevance for Melanoma Treatment. Cancers, 12(9), 2508. https://doi.org/10.3390/cancers12092508