The Origin and Immune Recognition of Tumor-Specific Antigens



Abstract
:Simple Summary
Abstract

1. Introduction
2. Misconceptions about TSAs
2.1. Neoantigens and the Fallacy of the Converse
2.2. Can mTSAs Be Identified without Mass Spectrometry Analyses?
2.2.1. Mass Spectrometry Validation of Predicted mTSAs
2.2.2. In-Depth Genomic Analyses
2.2.3. Can Reverse Immunology Eliminate False Positive TSA Predictions?
3. Strategies for Mass Spectrometry-Based Identification of aeTSAs
4. Immune Recognition of TSAs
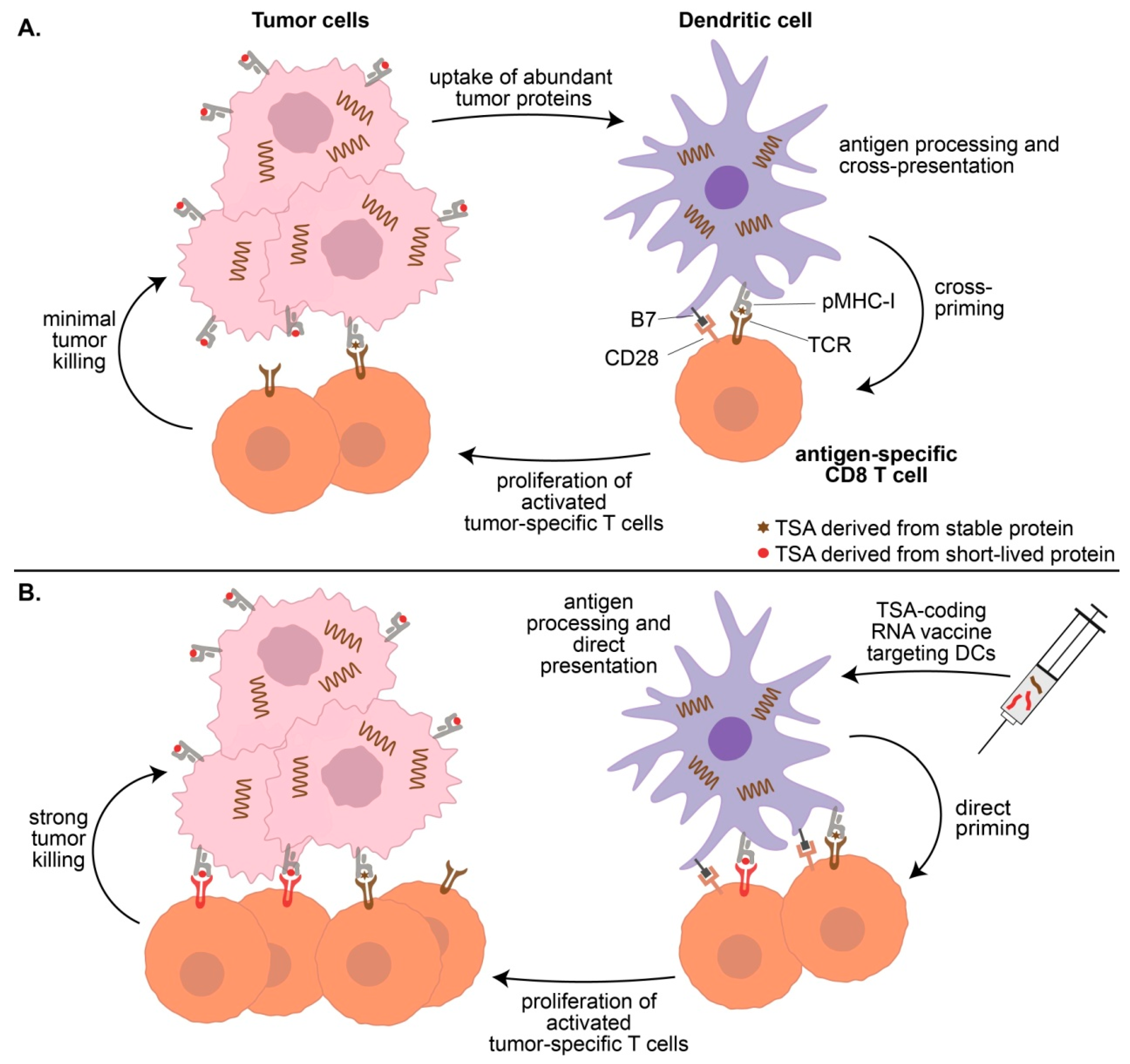
4.1. Cancer Cells Are Poor T-Cell Activators
4.2. Cross-Presentation Yields a Biased Representation of the TSA Repertoire
4.3. The Strength of Effector T-Cell Responses
4.4. Vaccination-Induced T-Cell Priming
4.5. Combining Vaccines and Immune Checkpoint Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Honjo, T. Combination therapy strategies for improving PD-1 blockade efficacy: A new era in cancer immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Haen, S.P.; Loffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Reustle, A.; Di Marco, M.; Meyerhoff, C.; Nelde, A.; Walz, J.S.; Winter, S.; Kandabarau, S.; Buttner, F.; Haag, M.; Backert, L.; et al. Integrative -omics and HLA-ligandomics analysis to identify novel drug targets for ccRCC immunotherapy. Genome Med. 2020, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Schuster, H.; Peper, J.K.; Bosmuller, H.C.; Rohle, K.; Backert, L.; Bilich, T.; Ney, B.; Loffler, M.W.; Kowalewski, D.J.; Trautwein, N.; et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA 2017, 114, E9942–E9951. [Google Scholar] [CrossRef] [Green Version]
- Shraibman, B.; Barnea, E.; Kadosh, D.M.; Haimovich, Y.; Slobodin, G.; Rosner, I.; Lopez-Larrea, C.; Hilf, N.; Kuttruff, S.; Song, C.; et al. Identification of Tumor Antigens Among the HLA Peptidomes of Glioblastoma Tumors and Plasma. Mol. Cell. Proteom. 2019, 18, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Loffler, M.W.; Kowalewski, D.J.; Backert, L.; Bernhardt, J.; Adam, P.; Schuster, H.; Dengler, F.; Backes, D.; Kopp, H.G.; Beckert, S.; et al. Mapping the HLA Ligandome of Colorectal Cancer Reveals an Imprint of Malignant Cell Transformation. Cancer Res. 2018, 78, 4627–4641. [Google Scholar] [CrossRef] [Green Version]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Mühlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Braunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma—A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Côté, C.; et al. Non-coding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Laverdure, J.P.; Lanoix, J.; Durette, C.; Coté, C.; Bonneil, E.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol. Res. 2020, 8, 544–555. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Wei, L.H.; Guo, J.U. Coding functions of “noncoding” RNAs. Science 2020, 367, 1074–1075. [Google Scholar] [CrossRef]
- Chen, J.; Brunner, A.D.; Cogan, J.Z.; Nunez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; Leonetti, M.D.; et al. Pervasive functional translation of noncanonical human open reading frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, E.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef]
- Larouche, J.D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.P.; Bonneil, E.; et al. Widespread and tissue-specific expression of endogenous retroelements in human somatic tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef]
- Koster, J.; Plasterk, R.H.A. A library of Neo Open Reading Frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci. Rep. 2019, 9, 6577. [Google Scholar] [CrossRef]
- Schmidt, M.; Lill, J.R. MHC class I presented antigens from malignancies: A perspective on analytical characterization & immunogenicity. J. Proteom. 2018. [Google Scholar] [CrossRef]
- Hardy, M.P.; Vincent, K.; Perreault, C. The Genomic Landscape of Antigenic Targets for T Cell-Based Leukemia Immunotherapy. Front. Immunol. 2019, 10, 2934. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Kubiniok, P.; Kovalchik, K.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A roadmap to predict and treat immune diseases. Mol. Cell. Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Van den Eynden, J.; Jimenez-Sanchez, A.; Miller, M.L.; Larsson, E. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet. 2019, 51, 1741–1748. [Google Scholar] [CrossRef]
- Alkallas, R.; Lajoie, M.; Hoang, K.V.; Lefrancois, P.; Lingrand, M.; Ahanfeshar-Adams, M.; Watters, K.; Spatz, A.; Zippin, J.H.; Najafabadi, H.S.; et al. Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nat. Cancer 2020, 1, 635–652. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Pearson, H.; Daouda, T.; Granados, D.P.; Durette, C.; Bonneil, E.; Courcelles, M.; Rodenbrock, A.; Laverdure, J.P.; Cote, C.; Mader, S.; et al. MHC class I-associated peptides derive from selective regions of the human genome. J. Clin. Investig. 2016, 126, 4690–4701. [Google Scholar] [CrossRef]
- Editorial, N.B. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [Google Scholar] [CrossRef]
- Granados, D.P.; Sriranganadane, D.; Daouda, T.; Zieger, A.; Laumont, C.M.; Caron-Lizotte, O.; Boucher, G.; Hardy, M.P.; Gendron, P.; Cote, C.; et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat. Commun. 2014, 5, 3600. [Google Scholar] [CrossRef]
- Dutoit, V.; Rubio-Godoy, V.; Pittet, M.J.; Zippelius, A.; Dietrich, P.Y.; Legal, F.A.; Guillaume, P.; Romero, P.; Cerottini, J.C.; Houghten, R.A.; et al. Degeneracy of antigen recognition as the molecular basis for the high frequency of naive A2/Melan-a peptide multimer(+) CD8(+) T cells in humans. J. Exp. Med. 2002, 196, 207–216. [Google Scholar] [CrossRef]
- Hesnard, L.; Legoux, F.; Gautreau, L.; Moyon, M.; Baron, O.; Devilder, M.C.; Bonneville, M.; Saulquin, X. Role of the MHC restriction during maturation of antigen-specific human T cells in the thymus. Eur. J. Immunol. 2016, 46, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Yotnda, P.; Garcia, F.; Peuchmaur, M.; Grandchamp, B.; Duval, M.; Lemonnier, F.; Vilmer, E.; Langlade-Demoyen, P. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J. Clin. Investig. 1998, 102, 455–462. [Google Scholar] [CrossRef]
- Popovic, J.; Li, L.P.; Kloetzel, P.M.; Leisegang, M.; Uckert, W.; Blankenstein, T. The only proposed T-cell epitope derived from the TEL-AML1 translocation is not naturally processed. Blood 2011, 118, 946–954. [Google Scholar] [CrossRef]
- Sewell, A.K. Why must T cells be cross-reactive? Nat. Rev. Immunol 2012, 12, 669–677. [Google Scholar] [CrossRef]
- Khosravi-Maharlooei, M.; Obradovic, A.; Misra, A.; Motwani, K.; Holzl, M.; Seay, H.R.; DeWolf, S.; Nauman, G.; Danzl, N.; Li, H.; et al. Crossreactive public TCR sequences undergo positive selection in the human thymic repertoire. J. Clin. Investig. 2019, 129, 2446–2462. [Google Scholar] [CrossRef] [Green Version]
- Wooldridge, L.; Ekeruche-Makinde, J.; van den Berg, H.A.; Skowera, A.; Miles, J.J.; Tan, M.P.; Dolton, G.; Clement, M.; Llewellyn-Lacey, S.; Price, D.A.; et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 2012, 287, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Probst, P.; Kopp, J.; Oxenius, A.; Colombo, M.P.; Ritz, D.; Fugmann, T.; Neri, D. Sarcoma Eradication by Doxorubicin and Targeted TNF Relies upon CD8+ T-cell Recognition of a Retroviral Antigen. Cancer Res. 2017, 77, 3644–3654. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Perreault, C. Exploiting non-canonical translation to identify new targets for T-cell based cancer immunotherapy. Cell. Mol Life Sci. 2017, 75, 607–621. [Google Scholar] [CrossRef]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Boegel, S.; Lower, M.; Bukur, T.; Sorn, P.; Castle, J.C.; Sahin, U. HLA and proteasome expression body map. BMC Med. Genom. 2018, 11, 36. [Google Scholar] [CrossRef]
- Benhammadi, M.; Mathé, J.; Dumont-Lagace, M.; Kobayashi, K.S.; Gaboury, L.; Brochu, S.; Perreault, C. IFN lambda enhances constitutive expression of MHC class I molecules on thymic epithelial cells. J. Immunol. 2020, 205, 1268–1280. [Google Scholar] [CrossRef]
- Chong, C.; Muller, M.; Pak, H.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [Green Version]
- Reuben, A.; Zhang, J.; Chiou, S.H.; Gittelman, R.M.; Li, J.; Lee, W.C.; Fujimoto, J.; Behrens, C.; Liu, X.; Wang, F.; et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020, 11, 603. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef] [Green Version]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Loh, P.R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8342 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Papula, A.L.; Poon, G.Y.P.; Wong, W.H.; Young, A.L.; Druley, T.E.; Fisher, D.S.; Blundell, J.R. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020, 367, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Kadouri, N.; Nevo, S.; Goldfarb, Y.; Abramson, J. Thymic epithelial cell heterogeneity: TEC by TEC. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Sansom, S.N.; Shikama-Dorn, N.; Zhanybekova, S.; Nusspaumer, G.; Macaulay, I.C.; Deadman, M.E.; Heger, A.; Ponting, C.P.; Hollander, G.A. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 2014, 24, 1918–1931. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, C.; Brochu, S.; Vanegas, J.R.; Dumont-Lagace, M.; Lemieux, S.; Perreault, C. Transcriptome sequencing of neonatal thymic epithelial cells. Sci. Rep. 2013, 3, 1860. [Google Scholar] [CrossRef] [Green Version]
- Pfammatter, S.; Bonneil, E.; Lanoix, J.; Vincent, K.; Hardy, M.P.; Courcelles, M.; Perreault, C.; Thibault, P. Extending the Comprehensiveness of Immunopeptidome Analyses Using Isobaric Peptide Labeling. Anal. Chem. 2020, 92, 9194–9204. [Google Scholar] [CrossRef]
- Chen, L.; McGowan, P.; Ashe, S.; Johnston, J.; Li, Y.; Hellstrom, I.; Hellstrom, K.E. Tumor immunogenicity determines the effect of B7 costimulation on T cell-mediated tumor immunity. J. Exp. Med. 1994, 179, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochsenbein, A.F. Immunological ignorance of solid tumors. Springer Semin. Immunopathol. 2005, 27, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yewdell, J.W.; Holly, J. DRiPs get molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Cosma, G.L.; Lobby, J.L.; Fay, E.J.; Siciliano, N.A.; Langlois, R.A.; Eisenlohr, L.C. Kinetically distinct processing pathways diversify the CD8(+) T cell response to a single viral epitope. Proc. Natl. Acad. Sci. USA 2020, 117, 19399–19407. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferre, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Rock, K.L. Cellular protein is the source of cross-priming antigen in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 3035–3040. [Google Scholar] [CrossRef] [Green Version]
- Norbury, C.C.; Basta, S.; Donohue, K.B.; Tscharke, D.C.; Princiotta, M.F.; Berglund, P.; Gibbs, J.; Bennink, J.R.; Yewdell, J.W. CD8+ T cell cross-priming via transfer of proteasome substrates. Science 2004, 304, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yewdell, J.W. Designing CD8+ T cell vaccines: It’s not rocket science (yet). Curr. Opin. Immunol. 2010, 22, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef]
- Sengupta, D.; Graham, M.; Liu, X.; Cresswell, P. Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. EMBO J. 2019, 38, e99266. [Google Scholar] [CrossRef]
- Cosma, G.L.; Eisenlohr, L.C. Impact of epitope density on CD8(+) T cell development and function. Mol. Immunol. 2019, 113, 120–125. [Google Scholar] [CrossRef]
- Meunier, M.C.; Delisle, J.S.; Bergeron, J.; Rineau, V.; Baron, C.; Perreault, C. T cells targeted against a single minor histocompatibility antigen can cure solid tumors. Nat. Med. 2005, 11, 1222–1229. [Google Scholar] [CrossRef]
- Wu, T.; Guan, J.; Handel, A.; Tscharke, D.C.; Sidney, J.; Sette, A.; Wakim, L.M.; Sng, X.Y.X.; Thomas, P.G.; Croft, N.P.; et al. Quantification of epitope abundance reveals the effect of direct and cross-presentation on influenza CTL responses. Nat. Commun. 2019, 10, 2846. [Google Scholar] [CrossRef] [Green Version]
- Gejman, R.S.; Chang, A.Y.; Jones, H.F.; DiKun, K.; Hakimi, A.A.; Schietinger, A.; Scheinberg, D.A. Rejection of immunogenic tumor clones is limited by clonal fraction. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Leisegang, M.; Engels, B.; Schreiber, K.; Yew, P.Y.; Kiyotani, K.; Idel, C.; Arina, A.; Duraiswamy, J.; Weichselbaum, R.R.; Uckert, W.; et al. Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation. Clin. Cancer Res. 2016, 22, 2734–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Liu, R.B.; Schreiber, K.; Schreiber, H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J. Exp. Med. 2010, 207, 2469–2477. [Google Scholar] [CrossRef]
- Spiotto, M.T.; Rowley, D.A.; Schreiber, H. Bystander elimination of antigen loss variants in established tumors. Nat. Med. 2004, 10, 294–298. [Google Scholar] [CrossRef]
- Blankenstein, T. The role of tumor stroma in the interaction between tumor and immune system. Curr. Opin. Immunol. 2005, 17, 180–186. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Melief, C.J.M.; Welters, M.J.P.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Tjalma, W.A.A.; Denys, H.; van Poelgeest, M.I.E.; Nijman, H.W.; et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Shao, W.; Pedrioli, P.G.A.; Wolski, W.; Scurtescu, C.; Schmid, E.; Vizcaino, J.A.; Courcelles, M.; Schuster, H.; Kowalewski, D.; Marino, F.; et al. The SysteMHC Atlas project. Nucleic Acids Res. 2018, 46, D1237–D1247. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Feature | TAAs | mTSAs | aeTSAs |
|---|---|---|---|
| Cancer-specific | No | Yes | Yes |
| Mutation | No | Yes | No |
| Shared among tumors | Yes | No | Yes |
| Number per tumor | Medium-High | Very low | Medium-High |
| Selected studies containing MS analyses | [9,10,11,12] | [13,14,15,16,17] | [18,19] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apavaloaei, A.; Hardy, M.-P.; Thibault, P.; Perreault, C. The Origin and Immune Recognition of Tumor-Specific Antigens. Cancers 2020, 12, 2607. https://doi.org/10.3390/cancers12092607
Apavaloaei A, Hardy M-P, Thibault P, Perreault C. The Origin and Immune Recognition of Tumor-Specific Antigens. Cancers. 2020; 12(9):2607. https://doi.org/10.3390/cancers12092607
Chicago/Turabian StyleApavaloaei, Anca, Marie-Pierre Hardy, Pierre Thibault, and Claude Perreault. 2020. "The Origin and Immune Recognition of Tumor-Specific Antigens" Cancers 12, no. 9: 2607. https://doi.org/10.3390/cancers12092607