Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. MEN1 and MENIN
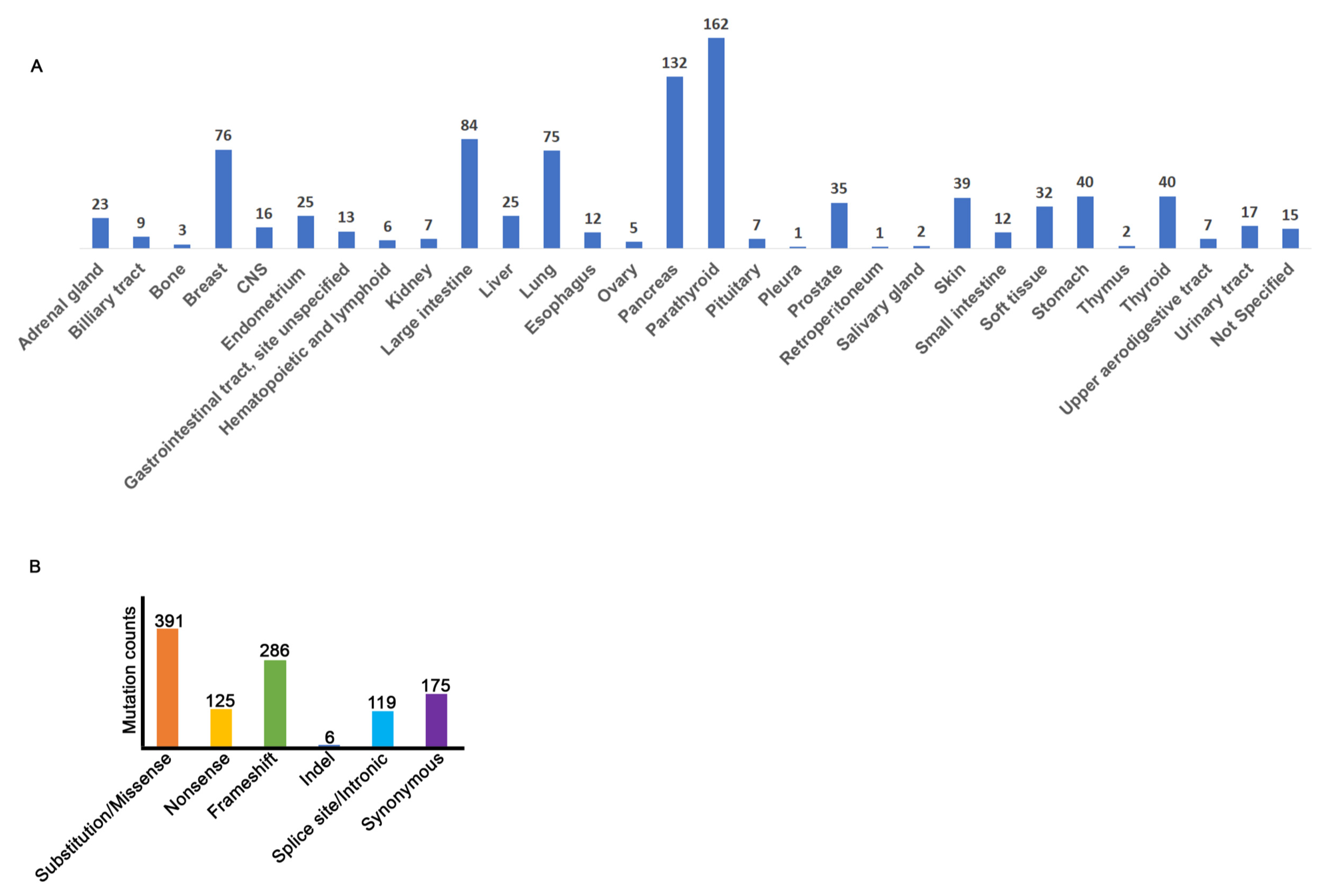
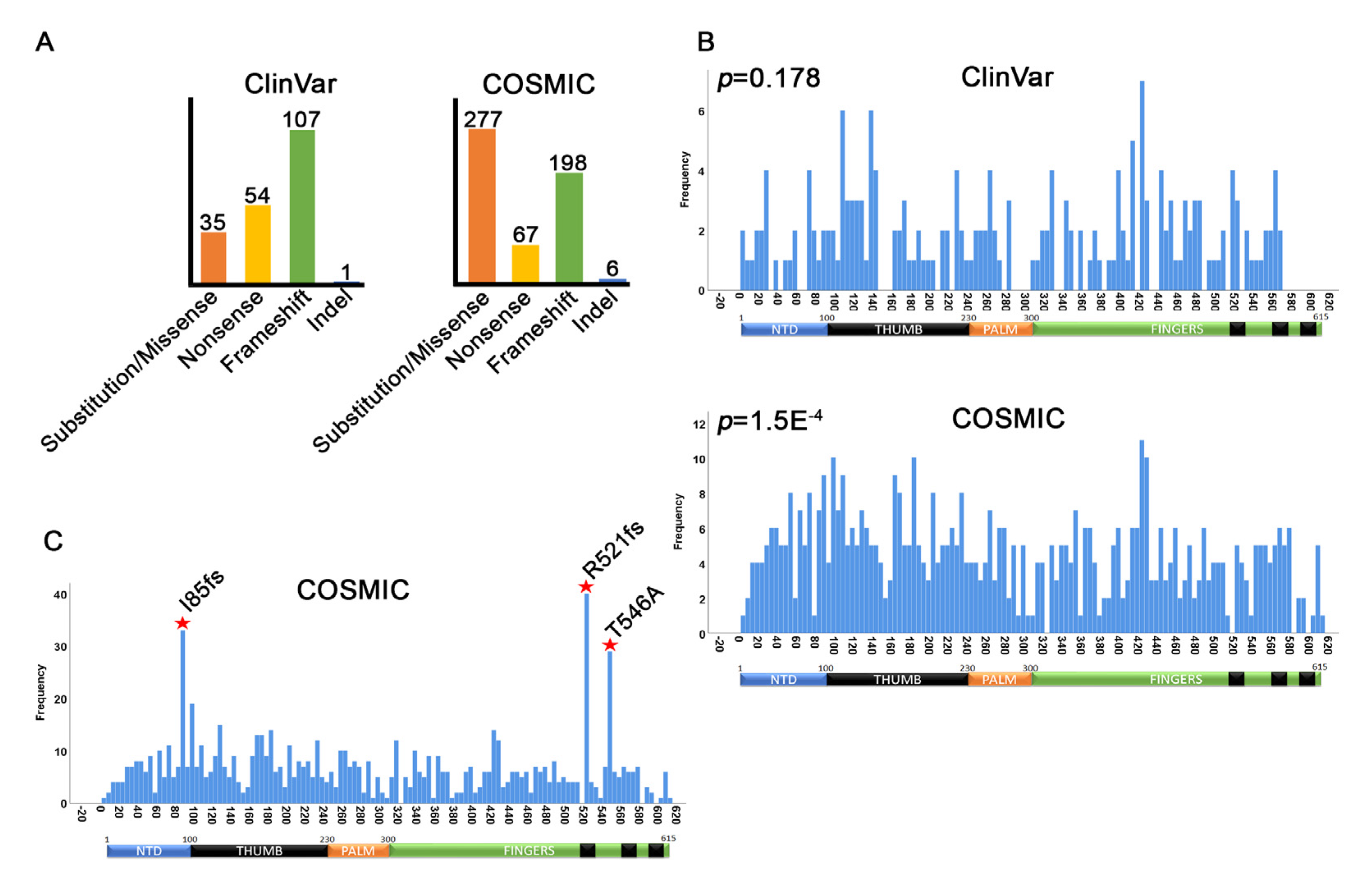
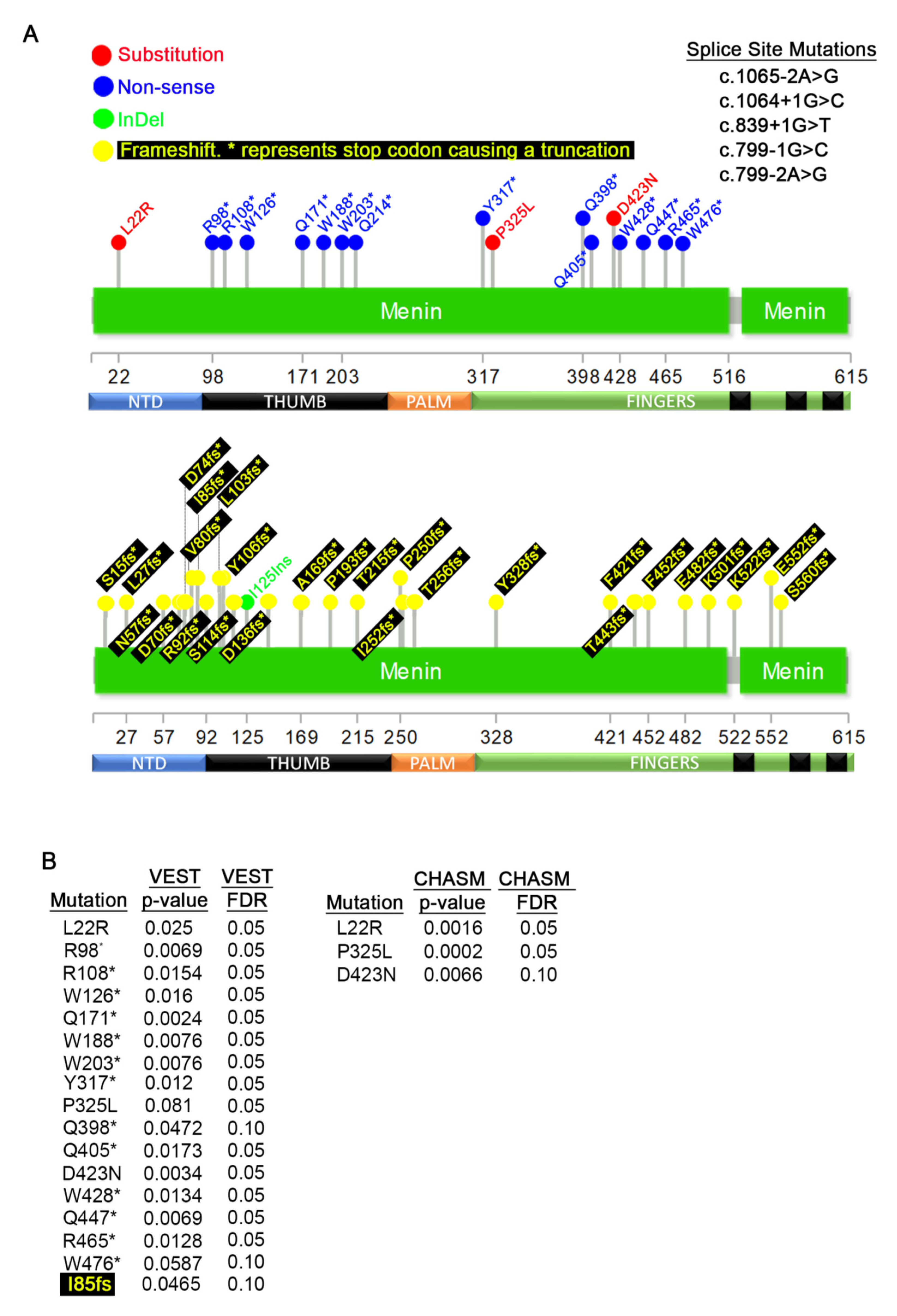
3.2. MEN1 Mutation Distribution
3.3. Germline MEN1 Mutations in Pancreatic, Parathyroid, Thyroid, Pituitary, and Thymus Tissues
3.4. Germline MEN1 Mutations Penetrant in other Cancers
3.5. MEN1 Mutations in Primary Tumors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, J.W.Y.; Hua, X.; Reidy-Lagunes, D.; Untch, B.R. MENIN loss as a tissue-specific driver of tumorigenesis. Mol. Cell. Endocrinol. 2018, 469, 98–106. [Google Scholar] [CrossRef]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef]
- Murai, M.J.; Chruszcz, M.; Reddy, G.; Grembecka, J.; Cierpicki, T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 2011, 286, 31742–31748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.R.; Tugendreich, S.; Hieter, P. Tetratrico peptide repeat interactions: To TPR or not to TPR? Trends Biochem. Sci. 1995, 20, 257–259. [Google Scholar] [CrossRef]
- Guru, S.C.; Goldsmith, P.K.; Burns, A.L.; Marx, S.J.; Spiegel, A.M.; Collins, F.S.; Chandrasekharappa, S.C. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. USA 1998, 95, 1630–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La, P.; Schnepp, R.W.; Clark, D.P.; Albert, C.S.; Hua, X. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology 2004, 145, 3443–3450. [Google Scholar] [CrossRef]
- La, P.; Desmond, A.; Hou, Z.; Silva, A.C.; Schnepp, R.W.; Hua, X. Tumor suppressor menin: The essential role of nuclear localization signal domains in coordinating gene expression. Oncogene 2006, 25, 3537–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Liu, R.; Jiang, X.; Lu, J.; Jiang, J.; Zhang, C.; Li, X.; Ning, G. Nuclear-cytoplasmic shuttling of menin regulates nuclear translocation of {beta}-catenin. Mol. Cell. Biol. 2009, 29, 5477–5487. [Google Scholar] [CrossRef] [Green Version]
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P.; et al. MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr. Relat. Cancer 2010, 17, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, T.; Nagamura, Y.; Ohkura, N. MEN1 gene and its mutations: Basic and clinical implications. Cancer Sci. 2009, 100, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Scacheri, P.C.; Davis, S.; Odom, D.T.; Crawford, G.E.; Perkins, S.; Halawi, M.J.; Agarwal, S.K.; Marx, S.J.; Spiegel, A.M.; Meltzer, P.S.; et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006, 2, e51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Song, T.Y.; Jung, K.Y.; Kim, S.G.; Cho, E.J. Direct interaction of menin leads to ubiquitin-proteasomal degradation of beta-catenin. Biochem. Biophys. Res. Commun. 2017, 492, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhao, H.; Yi, Y.; Nakata, Y.; Kalota, A.; Gewirtz, A.M. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Investig. 2010, 120, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, I.H.; Forsberg, L.; Pannett, A.A.; Meyen, E.; Piehl, F.; Turner, J.J.; Van de Ven, W.J.; Thakker, R.V.; Larsson, C.; Kas, K. Menin interacts directly with the homeobox-containing protein Pem. Biochem. Biophys. Res. Commun. 2001, 286, 426–431. [Google Scholar] [CrossRef]
- Sowa, H.; Kaji, H.; Hendy, G.N.; Canaff, L.; Komori, T.; Sugimoto, T.; Chihara, K. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J. Biol. Chem. 2004, 279, 40267–40275. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.K.; Guru, S.C.; Heppner, C.; Erdos, M.R.; Collins, R.M.; Park, S.Y.; Saggar, S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999, 96, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Heppner, C.; Bilimoria, K.Y.; Agarwal, S.K.; Kester, M.; Whitty, L.J.; Guru, S.C.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; Marx, S.J.; et al. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene 2001, 20, 4917–4925. [Google Scholar] [CrossRef] [Green Version]
- Gang, D.; Hongwei, H.; Hedai, L.; Ming, Z.; Qian, H.; Zhijun, L. The tumor suppressor protein menin inhibits NF-kappaB-mediated transactivation through recruitment of Sirt1 in hepatocellular carcinoma. Mol. Biol. Rep. 2013, 40, 2461–2466. [Google Scholar] [CrossRef]
- Shi, K.; Parekh, V.I.; Roy, S.; Desai, S.S.; Agarwal, S.K. The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic beta-cell proliferation and the expression of insulin regulators. Endocr. Relat. Cancer 2013, 20, 111–122. [Google Scholar] [CrossRef]
- Gurung, B.; Feng, Z.; Iwamoto, D.V.; Thiel, A.; Jin, G.; Fan, C.M.; Ng, J.M.; Curran, T.; Hua, X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013, 73, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, J.E.; Cho, E.J.; Liu, J.O.; Youn, H.D. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003, 63, 6135–6139. [Google Scholar] [PubMed]
- Wu, G.; Yuan, M.; Shen, S.; Ma, X.; Fang, J.; Zhu, L.; Sun, L.; Liu, Z.; He, X.; Huang, D.; et al. Menin enhances c-Myc-mediated transcription to promote cancer progression. Nat. Commun. 2017, 8, 15278. [Google Scholar] [CrossRef]
- Jin, S.; Mao, H.; Schnepp, R.W.; Sykes, S.M.; Silva, A.C.; D’Andrea, A.D.; Hua, X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003, 63, 4204–4210. [Google Scholar]
- Sukhodolets, K.E.; Hickman, A.B.; Agarwal, S.K.; Sukhodolets, M.V.; Obungu, V.H.; Novotny, E.A.; Crabtree, J.S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol. Cell. Biol. 2003, 23, 493–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.B.; Feng, Z.J.; Xu, B.; Wu, Y.; Yin, P.; Yang, Y.; Hua, X.; Jin, G.H. Suppression of lung adenocarcinoma through menin and polycomb gene-mediated repression of growth factor pleiotrophin. Oncogene 2009, 28, 4095–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef]
- McDonnell, J.E.; Gild, M.L.; Clifton-Bligh, R.J.; Robinson, B.G. Multiple endocrine neoplasia: An update. Intern. Med. J. 2019, 49, 954–961. [Google Scholar] [CrossRef]
- Larsson, C.; Skogseid, B.; Oberg, K.; Nakamura, Y.; Nordenskjold, M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988, 332, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Metz, D.C.; Jensen, R.T. Gastrointestinal neuroendocrine tumors: Pancreatic endocrine tumors. Gastroenterology 2008, 135, 1469–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, C.K.; Van Loon, K.; Bergsland, E.K.; Masharani, U.; Nakakura, E.K. Complicated Case Presentation: Management of Pancreatic Neuroendocrine Tumors in Multiple Endocrine Neoplasia Type 1. Pancreas 2017, 46, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Tatsi, C.; Stratakis, C.A. The Genetics of Pituitary Adenomas. J. Clin. Med. 2019, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Igarashi, H.; Uehara, H.; Berna, M.J.; Jensen, R.T. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: A Prospective study: Comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine 2013, 92, 135–181. [Google Scholar] [CrossRef]
- Agarwal, S.K. Multiple endocrine neoplasia type 1. Front. Horm Res. 2013, 41, 1–15. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Chen, Y.X.; Yan, J.; Keeshan, K.; Tubbs, A.T.; Wang, H.; Silva, A.; Brown, E.J.; Hess, J.L.; Pear, W.S.; Hua, X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Grembecka, J. Challenges and opportunities in targeting the menin-MLL interaction. Future Med. Chem. 2014, 6, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Bergamin, E.; Couture, J.F. The many facets of MLL1 regulation. Biopolymers 2013, 99, 136–145. [Google Scholar] [CrossRef]
- Uchino, S.; Noguchi, S.; Sato, M.; Yamashita, H.; Yamashita, H.; Watanabe, S.; Murakami, T.; Toda, M.; Ohshima, A.; Futata, T.; et al. Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res. 2000, 60, 5553–5557. [Google Scholar] [PubMed]
- Lemos, M.C.; Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008, 29, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, I.; Van de Ven, W.J.; Kas, K.; Zhang, C.X.; Giraud, S.; Wautot, V.; Buisson, N.; De Witte, K.; Salandre, J.; Lenoir, G.; et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 1997, 6, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Kester, M.B.; Debelenko, L.V.; Heppner, C.; Emmert-Buck, M.R.; Skarulis, M.C.; Doppman, J.L.; Kim, Y.S.; Lubensky, I.A.; Zhuang, Z.; et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum. Mol. Genet. 1997, 6, 1169–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front. Endocrinol. 2019, 10, 339. [Google Scholar] [CrossRef] [Green Version]
- Bergman, L.; Boothroyd, C.; Palmer, J.; Grimmond, S.; Walters, M.; Teh, B.; Shepherd, J.; Hartley, L.; Hayward, N. Identification of somatic mutations of the MEN1 gene in sporadic endocrine tumours. Br. J. Cancer 2000, 83, 1003–1008. [Google Scholar] [CrossRef]
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1). Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 355–370. [Google Scholar] [CrossRef]
- Machens, A.; Schaaf, L.; Karges, W.; Frank-Raue, K.; Bartsch, D.K.; Rothmund, M.; Schneyer, U.; Goretzki, P.; Raue, F.; Dralle, H. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): A multicentre study of 258 gene carriers. Clin. Endocrinol. 2007, 67, 613–622. [Google Scholar] [CrossRef]
- Marini, F.; Giusti, F.; Fossi, C.; Cioppi, F.; Cianferotti, L.; Masi, L.; Boaretto, F.; Zovato, S.; Cetani, F.; Colao, A.; et al. Multiple endocrine neoplasia type 1: Analysis of germline MEN1 mutations in the Italian multicenter MEN1 patient database. Endocrine 2018, 62, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Wautot, V.; Vercherat, C.; Lespinasse, J.; Chambe, B.; Lenoir, G.M.; Zhang, C.X.; Porchet, N.; Cordier, M.; Beroud, C.; Calender, A. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: Search for correlation between phenotype and the functional domains of the MEN1 protein. Hum. Mutat. 2002, 20, 35–47. [Google Scholar] [CrossRef]
- Kovesdi, A.; Toth, M.; Butz, H.; Szucs, N.; Sarman, B.; Pusztai, P.; Toke, J.; Reismann, P.; Faklya, M.; Toth, G.; et al. True MEN1 or phenocopy? Evidence for geno-phenotypic correlations in MEN1 syndrome. Endocrine 2019, 65, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Iacovazzo, D.; Hernandez-Ramirez, L.C.; Korbonits, M. Sporadic pituitary adenomas: The role of germline mutations and recommendations for genetic screening. Expert Rev. Endocrinol. Metab. 2017, 12, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, U.; Unuane, D.; Velkeniers, B.; Lissens, W.; Wuyts, W.; Bonduelle, M. A new double substitution mutation in the MEN1 gene: A limited penetrance and a specific phenotype. Eur. J. Hum. Genet. 2013, 21, 695–697. [Google Scholar] [CrossRef]
- Dreijerink, K.M.; van Beek, A.P.; Lentjes, E.G.; Post, J.G.; van der Luijt, R.B.; Canninga-van Dijk, M.R.; Lips, C.J. Acromegaly in a multiple endocrine neoplasia type 1 (MEN1) family with low penetrance of the disease. Eur. J. Endocrinol. 2005, 153, 741–746. [Google Scholar] [CrossRef]
- Weinhausel, A.; Kaserer, K.; Vierhapper, H.; Niederle, B.; Haas, O.A.; Study Group of Multiple Endocrine Neoplasia, A. Multiple endocrine neoplasia type 1 (MEN1) in Austria. Wien. Klin. Wochenschr. 2002, 114, 252–257. [Google Scholar] [PubMed]
- Giraud, S.; Zhang, C.X.; Serova-Sinilnikova, O.; Wautot, V.; Salandre, J.; Buisson, N.; Waterlot, C.; Bauters, C.; Porchet, N.; Aubert, J.P.; et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am. J. Hum. Genet. 1998, 63, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trump, D.; Farren, B.; Wooding, C.; Pang, J.T.; Besser, G.M.; Buchanan, K.D.; Edwards, C.R.; Heath, D.A.; Jackson, C.E.; Jansen, S.; et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM 1996, 89, 653–669. [Google Scholar] [CrossRef]
- Walls, G.V. Multiple endocrine neoplasia (MEN) syndromes. Semin. Pediatr. Surg. 2014, 23, 96–101. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Douville, C.; Carter, H.; Kim, R.; Niknafs, N.; Diekhans, M.; Stenson, P.D.; Cooper, D.N.; Ryan, M.; Karchin, R. CRAVAT: Cancer-related analysis of variants toolkit. Bioinformatics 2013, 29, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. S3). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrier, N.D. From Initial Description by Wermer to Present-Day MEN1: What have We Learned? World J. Surg. 2018, 42, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Khodaei-O’Brien, S.; Zablewska, B.; Fromaget, M.; Bylund, L.; Weber, G.; Gaudray, P. Heterogeneity at the 5’-end of MEN1 transcripts. Biochem. Biophys. Res. Commun. 2000, 276, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, L.; Zablewska, B.; Piehl, F.; Weber, G.; Lagercrantz, S.; Gaudray, P.; Hoog, C.; Larsson, C. Differential expression of multiple alternative spliceforms of the Men1 tumor suppressor gene in mouse. Int. J. Mol. Med. 2001, 8, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.; Hall, C.; Meng, F.; Lairmore, T.; Alpini, G.; Glaser, S. A Review of the Scaffold Protein Menin and its Role in Hepatobiliary Pathology. Gene Expr. 2017, 17, 251–263. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgleish, R.; Flicek, P.; Cunningham, F.; Astashyn, A.; Tully, R.E.; Proctor, G.; Chen, Y.; McLaren, W.M.; Larsson, P.; Vaughan, B.W.; et al. Locus Reference Genomic sequences: An improved basis for describing human DNA variants. Genome Med. 2010, 2, 24. [Google Scholar] [CrossRef]
- MacArthur, J.A.; Morales, J.; Tully, R.E.; Astashyn, A.; Gil, L.; Bruford, E.A.; Larsson, P.; Flicek, P.; Dalgleish, R.; Maglott, D.R.; et al. Locus Reference Genomic: Reference sequences for the reporting of clinically relevant sequence variants. Nucleic Acids Res. 2014, 42, D873–D878. [Google Scholar] [CrossRef]
- Marhuenda, Y.; Morales, D.; Pardo, M.C. A comparison of uniformity tests. Statistics 2005, 39, 315–327. [Google Scholar] [CrossRef]
- Falchetti, A. Genetics of multiple endocrine neoplasia type 1 syndrome: What’s new and what’s old. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Zhang, X.; Huang, X.; Yang, Y.; Hua, X. Regulation of cyclin B2 expression and cell cycle G2/m transition by menin. J. Biol. Chem. 2010, 285, 18291–18300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, A.B.; Xing, B.; Liu, C.; Naji, A.; Ma, X.; Simmons, R.A.; Hua, X. Menin and PRMT5 suppress GLP1 receptor transcript and PKA-mediated phosphorylation of FOXO1 and CREB. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E148–E166. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Yoshimoto, K.; Yamada, S.; Nishioka, H.; Ii, S.; Moritani, M.; Yamaoka, T.; Itakura, M. Absence of germ-line mutations of the multiple endocrine neoplasia type 1 (MEN1) gene in familial pituitary adenoma in contrast to MEN1 in Japanese. J. Clin. Endocrinol. Metab. 1998, 83, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, H.; Ohkura, N.; Takahashi, M.; Nagamura, Y.; Kitabayashi, I.; Tsukada, T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2004, 24, 6569–6580. [Google Scholar] [CrossRef] [Green Version]
- Heppner, C.; Kester, M.B.; Agarwal, S.K.; Debelenko, L.V.; Emmert-Buck, M.R.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Skarulis, M.C.; Doppman, J.L.; et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat. Genet. 1997, 16, 375–378. [Google Scholar] [CrossRef]
- Cinque, L.; Sparaneo, A.; Salcuni, A.S.; de Martino, D.; Battista, C.; Logoluso, F.; Palumbo, O.; Cocchi, R.; Maiello, E.; Graziano, P.; et al. MEN1 gene mutation with parathyroid carcinoma: First report of a familial case. Endocr. Connect. 2017, 6, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Kawai, T.; Saito, K.; Hishima, T.; Hayashi, Y.; Imura, J.; Hironaka, M.; Hosoya, Y.; Koike, M.; Fukayama, M. MEN1 gene mutations in sporadic neuroendocrine tumors of foregut derivation. Pathol. Int. 1999, 49, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Vortmeyer, A.O.; Pack, S.; Huang, S.; Pham, T.A.; Wang, C.; Park, W.S.; Agarwal, S.K.; Debelenko, L.V.; Kester, M.; et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997, 57, 4682–4686. [Google Scholar] [PubMed]
- Asteria, C.; Anagni, M.; Fugazzola, L.; Faglia, G.; Vezzadini, P.; Beck-Peccoz, P. MEN1 gene mutations are a rare event in patients with sporadic neuroendocrine tumors. Eur. J. Intern. Med. 2002, 13, 319–323. [Google Scholar] [CrossRef]
- Alvelos, M.I.; Vinagre, J.; Fonseca, E.; Barbosa, E.; Teixeira-Gomes, J.; Sobrinho-Simoes, M.; Soares, P. MEN1 intragenic deletions may represent the most prevalent somatic event in sporadic primary hyperparathyroidism. Eur. J. Endocrinol. 2013, 168, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Swarts, D.R.; Scarpa, A.; Corbo, V.; Van Criekinge, W.; van Engeland, M.; Gatti, G.; Henfling, M.E.; Papotti, M.; Perren, A.; Ramaekers, F.C.; et al. MEN1 gene mutation and reduced expression are associated with poor prognosis in pulmonary carcinoids. J. Clin. Endocrinol. Metab. 2014, 99, E374–E378. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.H.; Ebrahimi, S.A.; Wu, A.Y.; Kashefi, C.; Passaro, E., Jr.; Sawicki, M.P. Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Res. 1998, 58, 4417–4420. [Google Scholar]
- Totoki, Y.; Yoshida, A.; Hosoda, F.; Nakamura, H.; Hama, N.; Ogura, K.; Yoshida, A.; Fujiwara, T.; Arai, Y.; Toguchida, J.; et al. Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res. 2014, 24, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Hua, X.; Zhu, B.; Ravichandran, S.; Wang, M.; Nguyen, C.; Brodie, S.A.; Palleschi, A.; Alloisio, M.; Pariscenti, G.; et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002162. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Uchino, S.; Noguchi, S.; Nishioka, T.; Yamasaki, H.; Hashimoto, K.; Yoshimoto, K. Biallelic inactivation by somatic mutations of the MEN1 gene in sporadic parathyroid tumors. Cancer Lett. 2002, 175, 175–179. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretic, L.; Seidal, D.; Zander, T.; Leenders, F.; George, J.; Muller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5, 3518. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Rivals, E.; Jarne, P. DNA slippage occurs at microsatellite loci without minimal threshold length in humans: A comparative genomic approach. Genome Biol. Evol. 2010, 2, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Gay, L.M.; Wang, K.; Ali, S.M.; Chumsri, S.; Elvin, J.A.; Bose, R.; Vergilio, J.A.; Suh, J.; Yelensky, R.; et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: An emerging opportunity for anti-HER2 targeted therapies. Cancer 2016, 122, 2654–2662. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Sheehan, C.E.; Boguniewicz, A.B.; Otto, G.; Downing, S.R.; Sun, J.; He, J.; Curran, J.A.; Ali, S.; et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin. Cancer Res. 2013, 19, 2668–2676. [Google Scholar] [CrossRef] [Green Version]
- Lippert, J.; Appenzeller, S.; Liang, R.; Sbiera, S.; Kircher, S.; Altieri, B.; Nanda, I.; Weigand, I.; Gehrig, A.; Steinhauer, S.; et al. Targeted Molecular Analysis in Adrenocortical Carcinomas: A Strategy Toward Improved Personalized Prognostication. J. Clin. Endocrinol. Metab. 2018, 103, 4511–4523. [Google Scholar] [CrossRef]
- Cromer, M.K.; Starker, L.F.; Choi, M.; Udelsman, R.; Nelson-Williams, C.; Lifton, R.P.; Carling, T. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J. Clin. Endocrinol. Metab. 2012, 97, E1774–E1781. [Google Scholar] [CrossRef] [Green Version]
- Goebel, S.U.; Heppner, C.; Burns, A.L.; Marx, S.J.; Spiegel, A.M.; Zhuang, Z.; Lubensky, I.A.; Gibril, F.; Jensen, R.T.; Serrano, J. Genotype/phenotype correlation of multiple endocrine neoplasia type 1 gene mutations in sporadic gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Pardi, E.; Marcocci, C.; Borsari, S.; Saponaro, F.; Torregrossa, L.; Tancredi, M.; Raspini, B.; Basolo, F.; Cetani, F. Aryl hydrocarbon receptor interacting protein (AIP) mutations occur rarely in sporadic parathyroid adenomas. J. Clin. Endocrinol. Metab. 2013, 98, 2800–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cetani, F.; Pardi, E.; Giovannetti, A.; Cerrai, P.; Borsari, S.; Vignali, E.; Picone, A.; Cianferotti, L.; Miccoli, P.; Pinchera, A.; et al. Six novel MEN1 gene mutations in sporadic parathyroid tumors. Hum. Mutat. 2000, 16, 445. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, A.; Vincent-Salomon, A.; Pivot, X.; Sertier, A.S.; Thomas, E.; Tonon, L.; Boyault, S.; Mulugeta, E.; Treilleux, I.; MacGrogan, G.; et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat. Commun. 2016, 7, 12222. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.M.; Mengel, M.; Heinze, M.; Simon, D.; Scheuring, S.; Kohrer, K.; Roher, H.D. Complete sequencing and messenger ribonucleic acid expression analysis of the MEN I gene in adrenal cancer. J. Clin. Endocrinol. Metab. 2000, 85, 441–448. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Dutton-Regester, K.; Kakavand, H.; Aoude, L.G.; Stark, M.S.; Gartside, M.G.; Johansson, P.; O’Connor, L.; Lanagan, C.; Tembe, V.; Pupo, G.M.; et al. Melanomas of unknown primary have a mutation profile consistent with cutaneous sun-exposed melanoma. Pigment. Cell Melanoma Res. 2013, 26, 852–860. [Google Scholar] [CrossRef]
- Prostatic intraepithelial neoplasia: Significance and correlation with prostate-specific antigen and transrectal ultrasound. Proceedings of a workshop of the National Prostate Cancer Detection Project. March 13, 1989, Bethesda, Maryland. Urology 1989, 34, 2–69.
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Shimamoto, A.; Goto, M.; Furuichi, Y. Impaired nuclear localization of defective DNA helicases in Werner’s syndrome. Nat. Genet. 1997, 16, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Fang, X.; Ma, N.; Lin, Q.; Huang, Z.; Liu, W.; Xu, M.; Chen, X.; Zhang, W.; Zhang, Y. Myeloperoxidase-deficient zebrafish show an augmented inflammatory response to challenge with Candida albicans. Fish. Shellfish Immunol. 2015, 44, 109–116. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yang, C.; Li, X.; Luo, W.; Roy, B.; Xiong, T.; Zhang, X.; Yang, H.; Wang, J.; Ye, Z.; et al. The landscape of somatic mutation in sporadic Chinese colorectal cancer. Oncotarget 2018, 9, 27412–27422. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Chou, W.C.; Lin, P.H.; Yeh, Y.C.; Shyr, Y.M.; Fang, W.L.; Wang, S.E.; Liu, C.Y.; Chang, P.M.; Chen, M.H.; Hung, Y.P.; et al. Genes involved in angiogenesis and mTOR pathways are frequently mutated in Asian patients with pancreatic neuroendocrine tumors. Int. J. Biol. Sci. 2016, 12, 1523–1532. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.M.; Jung, S.H.; Kim, M.S.; Baek, I.P.; Park, S.W.; Lee, S.H.; Lee, H.H.; Kim, S.S.; Chung, Y.J.; Lee, S.H. The mutational burdens and evolutionary ages of early gastric cancers are comparable to those of advanced gastric cancers. J. Pathol. 2014, 234, 365–374. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Debelenko, L.V.; Brambilla, E.; Agarwal, S.K.; Swalwell, J.I.; Kester, M.B.; Lubensky, I.A.; Zhuang, Z.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum. Mol. Genet. 1997, 6, 2285–2290. [Google Scholar] [CrossRef] [PubMed]
- Veschi, S.; Lattanzio, R.; Aceto, G.M.; Curia, M.C.; Magnasco, S.; Angelucci, D.; Cama, A.; Piantelli, M.; Battista, P. Alterations of MEN1 and E-cadherin/beta-catenin complex in sporadic pulmonary carcinoids. Int. J. Oncol. 2012, 41, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzi, W.; Renon, M.; Vercherat, C.; Hamze, Z.; Lacheretz-Bernigaud, A.; Wang, H.; Blanc, M.; Roche, C.; Calender, A.; Chayvialle, J.A.; et al. MEN1 missense mutations impair sensitization to apoptosis induced by wild-type menin in endocrine pancreatic tumor cells. Gastroenterology 2008, 135, 1698–1709.e1692. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene or Region | MEN1 Coordinates (Amino Acids) | Function |
|---|---|---|
| MEN1 Domains | ||
| NTD | 1–100 | Crystal structure identified domain |
| Thumb | 101–1230 | Crystal structure identified domain |
| Palm | 231–1300 | Crystal structure identified domain |
| Fingers | 301–1305 | Crystal structure identified domain |
| NLS1 | 479–1497 | Nuclear localization sequence |
| NLS2 | 588–1608 | Nuclear localization sequence |
| NLS3 | 546–1672 | Nuclear localization sequence |
| NES1 | 33–151 | Nuclear exit sequence |
| NES2 | 253–1267 | Nuclear exit sequence |
| MEN1 Interacting Proteins | ||
| KMTA2 (MLL1) | ~230–1300 | Mixed lineage leukemia |
| LEDGF | ~1–1100 | Chromatin associated factor, required for MLL oncogenic transformation |
| JunD | 1–140, 323–1448 | Transcriptional activator, subunit of AP-1 transcription complex |
| HLXB9 | 41–1177, 323? | Homeobox gene involved in pancreas development, neural motor protein |
| SMAD3 | 41–1278, 477–1615 | TGFB signaling pathway, cell proliferation, transcriptional regulation |
| MYC | 360-615 (fingers), maybe NTD | Transcriptional regulation, cell cycle, apoptosis, cellular transformation |
| PEM (mouse) | 278–1476 | Homeobox gene, embryonic and placenta expression |
| NF-kB subunits | 305–1476 | Transcriptional regulators, inflammation, immune response, cell proliferation |
| SIRT1 | 305–1476 | Sirtuitin, gene silencing |
| SIN3A | 295–1450 | Gene expression regulator, embryogenesis, cell proliferation, senescence |
| HDAC1 | 295–1450 | Histone deacetylase, transcriptional regulator, cell proliferation and differentiation |
| PRMT5 | L22, A242 | Arginine methyltransferase, transcriptional regulation, DNA damage repair |
| FANCD2 | 219–1395 | Fancomi anemia complex subunit, DNA damage repair |
| RPA2 | 1–140, 286–1448 | Replication Protein A subunit |
| 1 Transcript Variant | Transcript Size (Bases) | Protein Isoform | Protein Size (Amino Acids) |
|---|---|---|---|
| 1 | 2785 | 2 1 | 615 |
| e1B | 2748 | 1 | 615 |
| e1C | 2736 | 1 | 615 |
| e1D | 3712 | 1 | 615 |
| e1E | 3179 | 1 | 615 |
| e1F1 | 3015 | 1 | 615 |
| 2 | 2770 | 3 2 | 610 |
| 3 | 2828 | 3 | 652 |
| 4 | 2712 | 2 | 610 |
| 5 | 2702 | 2 | 610 |
| 6 | 2960 | 2 | 610 |
| 7 | 2855 | 4 | 575 |
| 8 | 2609 | 4 | 575 |
| 4 MEN1-207 | 3150 | 2 | 610 |
| 5 MEN1-205 | 2868 | ? | 555 |
| X1 | 3629 | X1 | 657 |
| X2 | 3629 | 3 | 652 |
6 Isoform alignment schematic. | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelakurti, D.D.; Pappula, A.L.; Rajasekaran, S.; Miles, W.O.; Petreaca, R.C. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers 2020, 12, 2616. https://doi.org/10.3390/cancers12092616
Nelakurti DD, Pappula AL, Rajasekaran S, Miles WO, Petreaca RC. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers. 2020; 12(9):2616. https://doi.org/10.3390/cancers12092616
Chicago/Turabian StyleNelakurti, Devi D., Amrit L. Pappula, Swetha Rajasekaran, Wayne O. Miles, and Ruben C. Petreaca. 2020. "Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer" Cancers 12, no. 9: 2616. https://doi.org/10.3390/cancers12092616
APA StyleNelakurti, D. D., Pappula, A. L., Rajasekaran, S., Miles, W. O., & Petreaca, R. C. (2020). Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers, 12(9), 2616. https://doi.org/10.3390/cancers12092616