Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
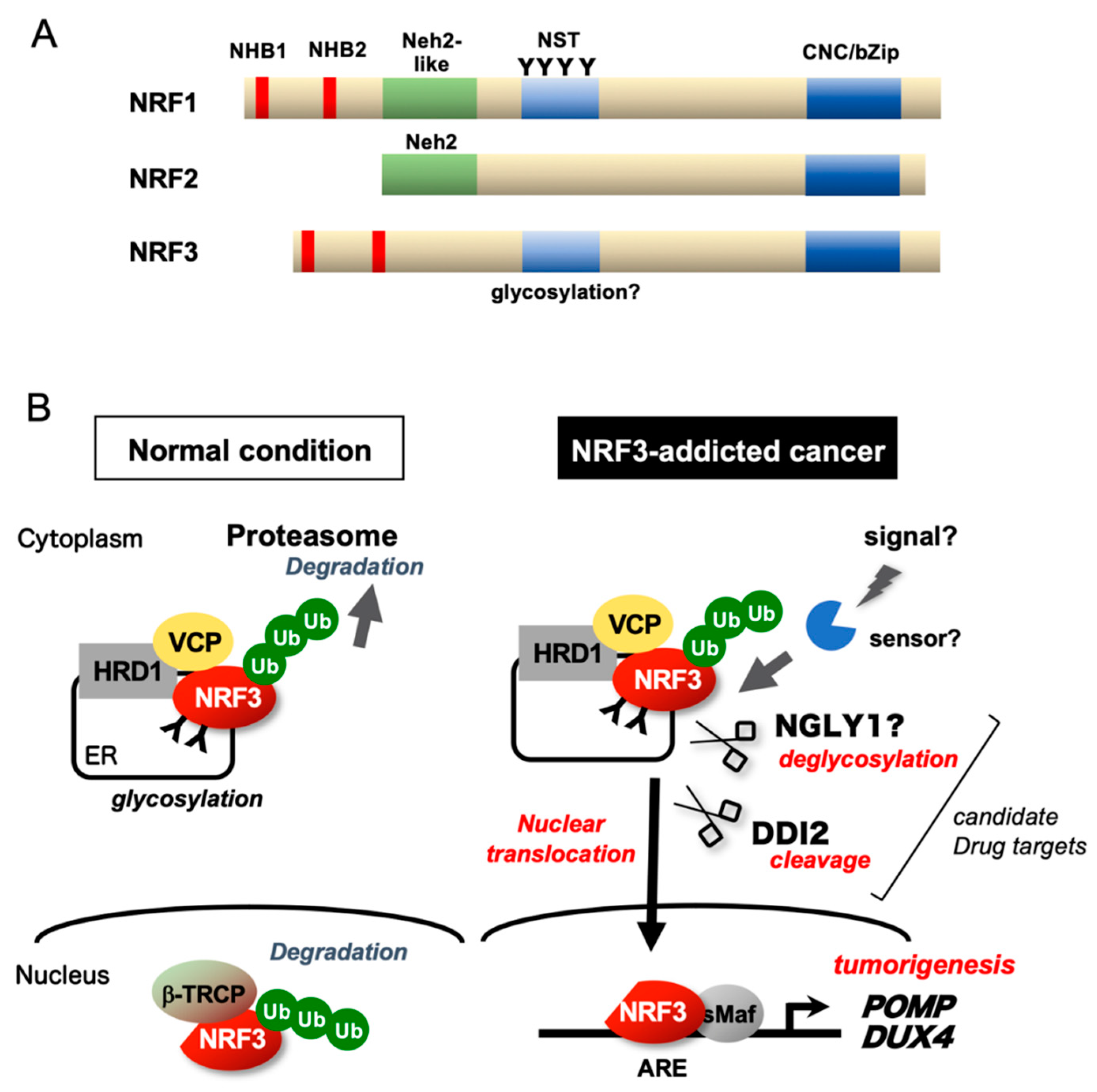{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Newly Discovered Addiction of Colon Cancer Cells to NRF3
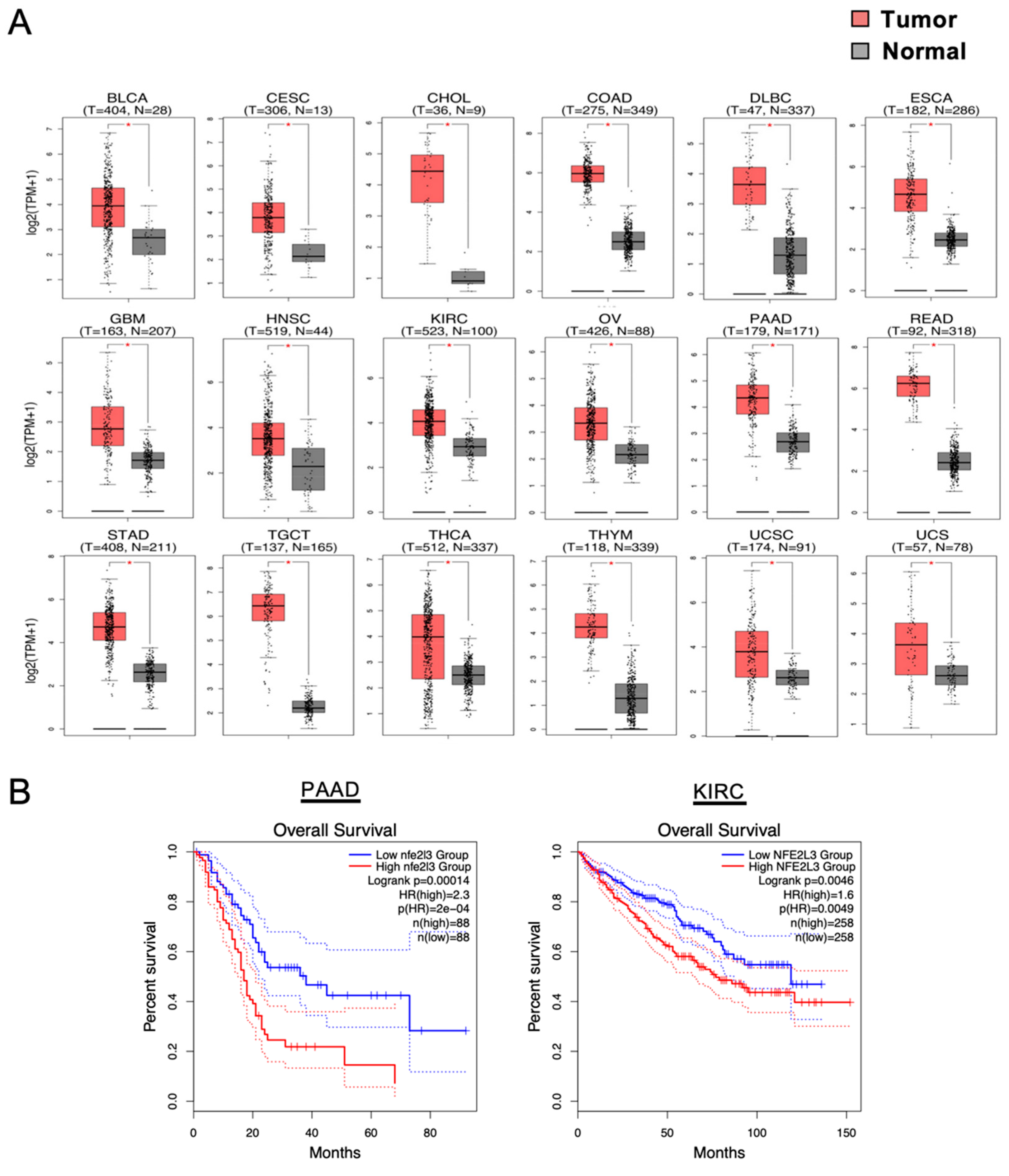
2.1. Remarkable NRF3 Induction Associated with Malignancy
2.2. Mechanisms of the NRF3 Induction in Colon Cancer
2.3. NRF3 Transcriptome in Cancer Cells
2.4. Cancer Cells are Highly Addicted to Proteostasis
2.5. Gene Regulation and Assembly Mechanisms of the Proteasomes
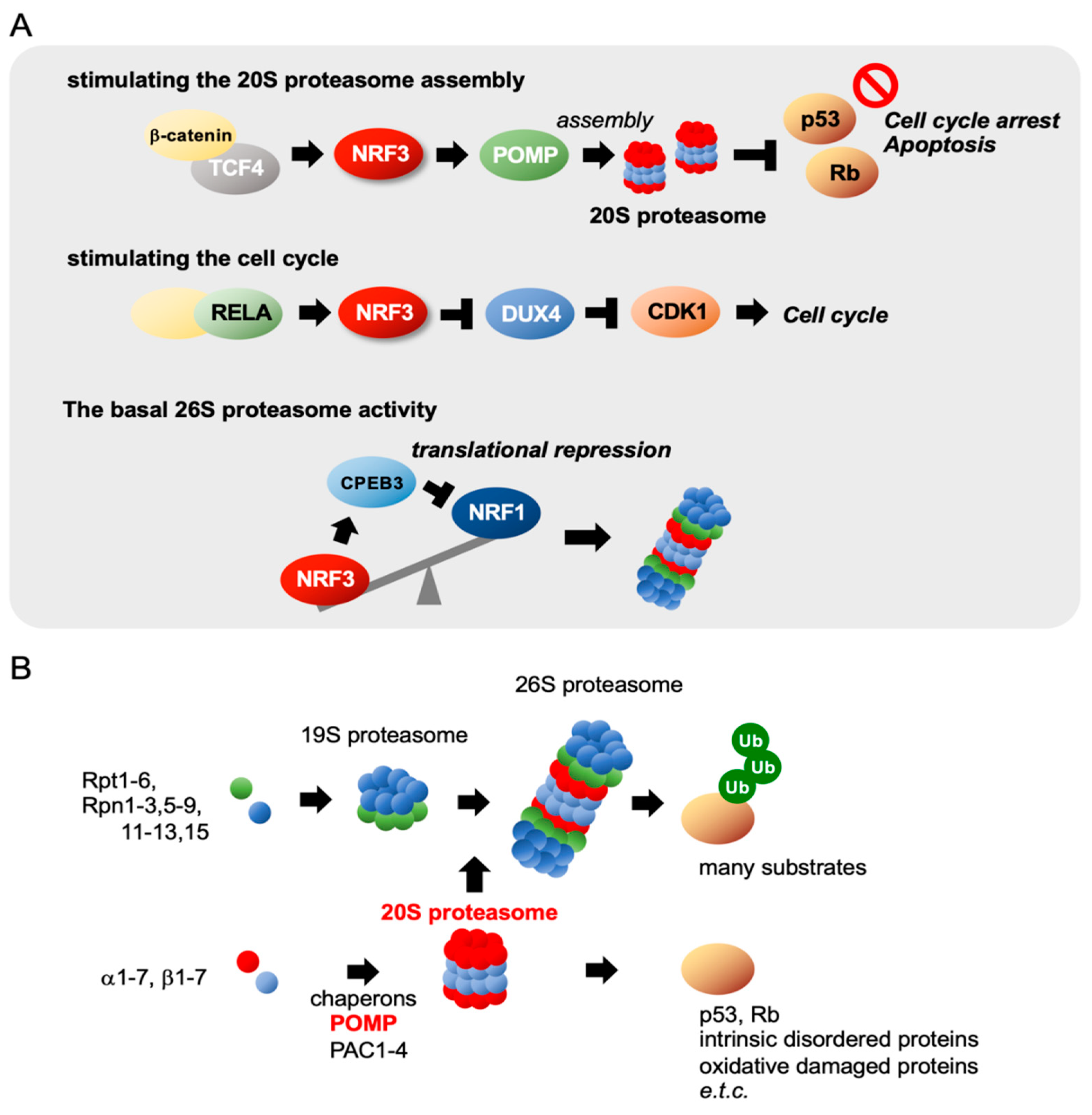
2.6. NRF3 Stimulates 20S Proteasome Assembly and the Cell Cycle
2.7. Colon Cancer Cells May Favor NRF3 for Proteostasiss
NRF3 Suppresses NRF1 Translation by Inducing CPEB3
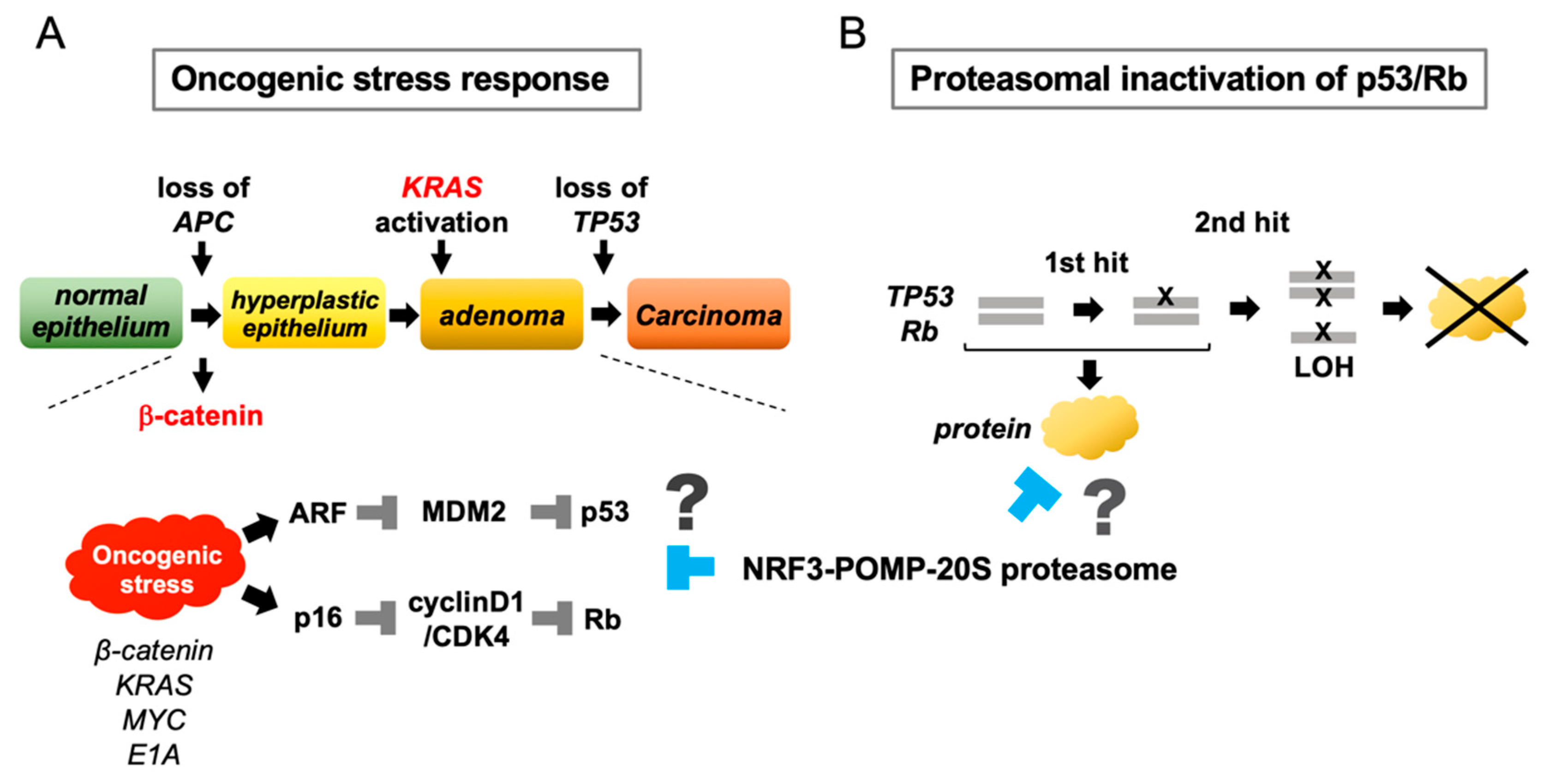
2.8. Conceptual Hypotheses: The Oncogenic Stress Response Hypothesis and the Proteasomal Inactivation of Tumor Suppressors Hypothesis
2.9. Remaining Issues Regarding the NRF3–POMP–20S Proteasome Axis
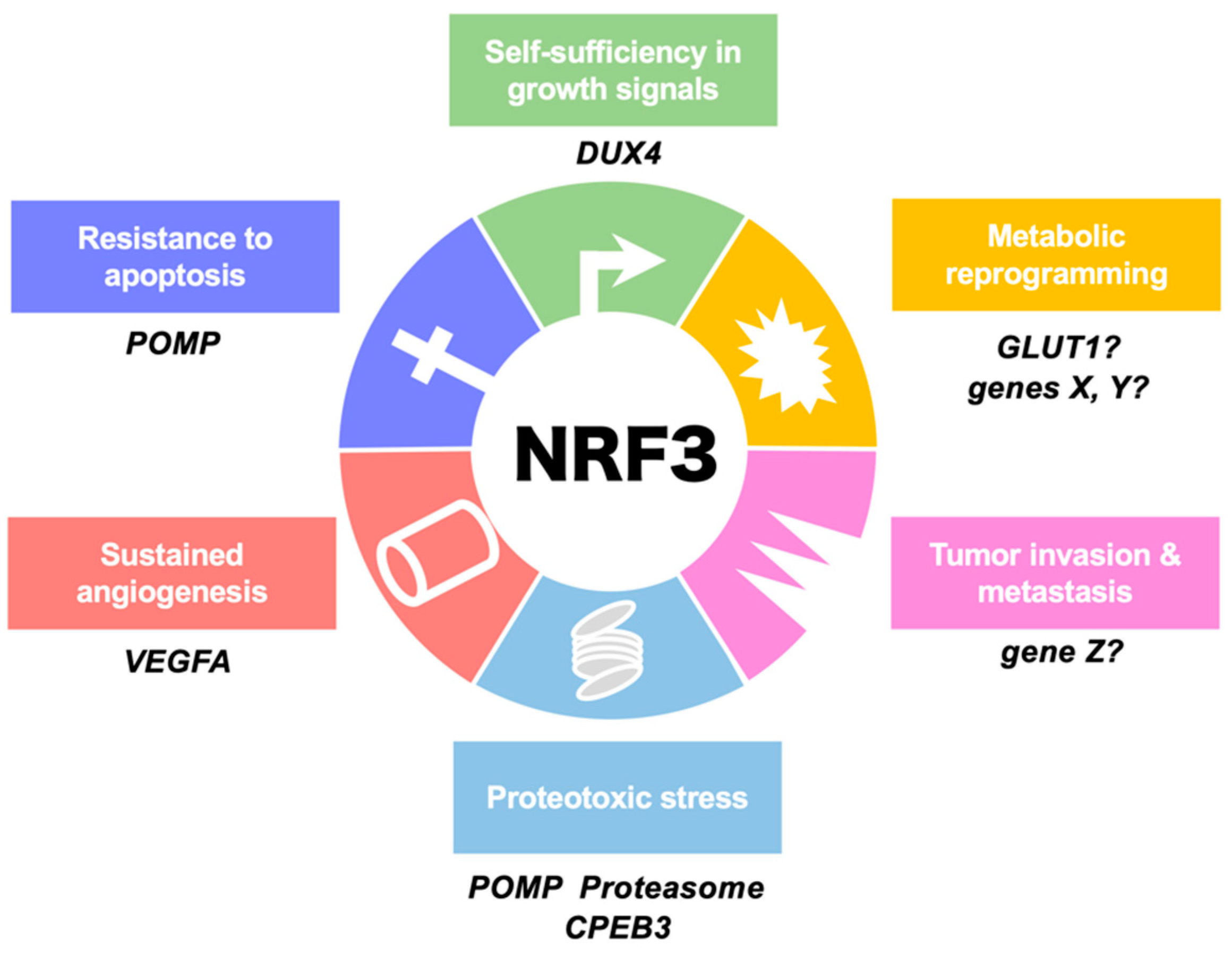
3. NRF3-Driven Cancer Malignancy
4. NRF3 as the Tumor Suppressor
5. Differences between NRF3 and NRF2
5.1. Phenotypes of Gene Targeting Mice
5.2. Protein Structures and Regulatory Mechanisms
5.3. Missense Mutations in Cancer
5.4. Functional Redundancy between NRF3 and NRF1
6. Cross Talk of NRF3 with Other Transcription Factors
7. Development of New Anticancer Drugs that Suppress NRF3
8. An Emerging Concern: The Authenticity of Commercial Anti-NRF3 Antibodies
9. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Review Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Ito, E.; Toki, T.; Kogame, K.; Takahashi, S.; Igarashi, K.; Hayashi, N.; Yamamoto, M. Molecular Cloning and Functional Characterization of a New Cap ’n’ Collar Family Transcription Factor Nrf3. J. Biol. Chem. 1999, 274, 6443–6452. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC Transcription Factors as Revealed by Gene Targeting of the Nrf3 Locus. Mol. Cell. Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef] [Green Version]
- Chevillard, G.G.; Nouhi, Z.; Anna, D.; Paquet, M.M.; Blank, V. Nrf3-deficient mice are not protected against acute lung and adipose tissue damages induced by butylated hydroxytoluene. FEBS Lett. 2010, 584, 923–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aono, S.; Hatanaka, A.; Hatanaka, A.; Gao, Y.; Hippo, Y.; Taketo, M.M.; Waku, T.; Kobayashi, A. β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3344. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Waku, T. New addiction to the NRF2-related factor NRF3 in cancer cells: Ubiquitin-independent proteolysis through the 20S proteasome. Cancer Sci. 2020, 111, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 721–735. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, H.; Dong, H.; Zhu, L.; Wang, S.; Wang, P.; Ren, Q.; Zhu, H.; Chen, J.; Lin, Z.; et al. LAT, HOXD3 and NFE2L3 identified as novel DNA methylation-driven genes and prognostic markers in human clear cell renal cell carcinoma by integrative bioinformatics approaches. J. Cancer 2019, 10, 6726–6737. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Hu, X. Identification and Comprehensive Validation of a DNA Methylation-Driven Gene-Based Prognostic Model for Clear Cell Renal Cell Carcinoma. DNA Cell Biol. 2020. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Waku, T.; Nakamura, N.; Koji, M.; Watanabe, H.; Katoh, H.; Tatsumi, C.; Tamura, N.; Hatanaka, A.; Hirose, S.; Katayama, H.; et al. NRF3-POMP-20S proteasome assembly axis promotes cancer development via ubiquitin-independent proteolysis of p53 and Rb. Mol. Cell. Biol. 2020, 40, e00597-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bury, M.; Le Calvé, B.; Lessard, F.; Dal Maso, T.; Saliba, J.; Michiels, C.; Ferbeyre, G.; Blank, V. NFE2L3 Controls Colon Cancer Cell Growth through Regulation of DUX4, a CDK1 Inhibitor. Cell Rep. 2019, 29, 1469–1481. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harb. Symp. Quant. Biol. 2012, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Ann. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Waku, T.; Katayama, H.; Hiraoka, M.; Hatanaka, A.; Nakamura, N.; Tanaka, Y.; Tamura, N.; Watanabe, A.; Kobayashi, A. NFE2L1 and NFE2L3 complementarily maintain basal proteasome activity in cancer cells through CPEB3-mediated translational repression. Mol. Cell. Biol. 2020, 40, 1–13. [Google Scholar] [CrossRef]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]
- Hamazaki, J.; Murata, S. Er-resident transcription factor NRF1 regulates proteasome expression and beyond. Int. J. Mol. Sci. 2020, 21, 3683. [Google Scholar] [CrossRef]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Chevillard, G.; Blank, V. NFE2L3 (NRF3): The Cinderella of the Cap’n’Collar transcription factors. Cell. Mol. Life Sci. 2011, 68, 3337–3348. [Google Scholar] [CrossRef]
- Chénais, B.; Derjuga, A.; Massrieh, W.; Red-Horse, K.; Bellingard, V.; Fisher, S.J.; Blank, V.; Chenais, B.; Derjuga, A.; Massrieh, W.; et al. Functional and placental expression analysis of the human NRF3 transcription factor. Mol. Endocrinol. 2005, 19, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J. Biol. Chem. 2019, 294, 18131–18149. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.M.M.A.; Katoh, H.; Hatanaka, A.; Iwanari, H.; Nakamura, N.; Hamakubo, T.; Natsume, T.; Waku, T.; Kobayashi, A. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci. Rep. 2017, 7, 12494. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutsadakis, I.A. Proteasome expression and activity in cancer and cancer stem cells. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal Degradation Is Transcriptionally Controlled by TCF11 via an ERAD-Dependent Feedback Loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Taniguchi, H.; Ito, Y.; Morita, T.; Karim, M.R.R.; Ohtake, N.; Fukagai, K.; Ito, T.; Okamuro, S.; Iemura, S.-I.; et al. The casein kinase 2-nrf1 axis controls the clearance of ubiquitinated proteins by regulating proteasome gene expression. Mol. Cell. Biol. 2013, 33, 3461–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, Z.; Goldberg, A.L. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Schulz, R.; Edmunds, S.; Krüger, E.; Markert, E.; Gaedcke, J.; Cormet-Boyaka, E.; Ghadimi, M.; Beissbarth, T.; Levine, A.J.; et al. MicroRNA-101 Suppresses Tumor Cell Proliferation by Acting as an Endogenous Proteasome Inhibitor via Targeting the Proteasome Assembly Factor POMP. Mol. Cell 2015, 59, 243–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–16. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. Regulation of the Expression of Cytoplasmic Polyadenylation Element Binding Proteins for the Treatment of Cancer. Anticancer Res. 2016, 36, 5673–5680. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrogio, A.; Nagaoka, K.; Richter, J.D. Translational control of cell growth and malignancy by the CPEBs. Nat. Rev. Cancer 2013, 13, 283–290. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, K.M.; Sweet-Cordero, A. New insights into oncogenic stress. Nat. Genet. 2011, 43, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van De Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and targeting of TP53 pathway in multiple myeloma. Front. Oncol. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19(Arf), interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Sharpless, N.E. The Regulation of INK4/ARF in Cancer and Aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Pauklin, S.; Kristjuhan, A.; Maimets, T.; Jaks, V. ARF and ATM/ATR cooperate in p53-mediated apoptosis upon oncogenic stress. Biochem. Biophys. Res. Commun. 2005, 334, 386–394. [Google Scholar] [CrossRef]
- Payne, S.R.; Kemp, C.J. Tumor suppressor genetics. Carcinogenesis 2005, 26, 2031–2045. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The Contribution of the 20S Proteasome to Proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-nissan, G.; Sharon, M. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Wang, C.; Saji, M.; Justiniano, S.E.; Yusof, A.M.; Zhang, X.; Yu, L.; Fernández, S.; Wakely, P., Jr.; La Perle, K.; Nakanishi, H.; et al. RCAN1-4 is a thyroid cancer growth and metastasis suppressor. J. Clin. Investig. Insight 2017, 2, 1–15. [Google Scholar] [CrossRef]
- Yu, M.M.; Feng, Y.H.; Zheng, L.; Zhang, J.; Luo, G.H. Short hairpin RNA-mediated knockdown of nuclear factor erythroid 2-like 3 exhibits tumor-suppressing effects in hepatocellular carcinoma cells. World J. Gastroenterol. 2019, 25, 1210–1223. [Google Scholar] [CrossRef]
- Wang, H.; Zhan, M.; Yang, R.; Shi, Y.; Liu, Q.; Wang, J. Elevated expression of NFE2L3 predicts the poor prognosis of pancreatic cancer patients. Cell Cycle 2018, 17, 2164–2174. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, Q.; Wang, L.; Wang, H.; Yin, F. A DNA methylation signature to improve survival prediction of gastric cancer. Clin. Epigenetics 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Rauscher, G.H.; Kresovich, J.K.; Poulin, M.; Yan, L.; Macias, V.; Mahmoud, A.M.; Al-Alem, U.; Kajdacsy-Balla, A.; Wiley, E.L.; Tonetti, D.; et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer 2015, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zheng, Z.; Chen, Q.; Pan, Y.; Lu, H.; Zhang, H.; Yu, Y.; Dai, Y. NRF3 suppresses breast cancer cell metastasis and cell proliferation and is a favorable predictor of survival in breast cancer. Onco. Targets. Ther. 2019, 12, 3019–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Li, Y.; Lu, J.; Chen, C.; Wang, W.; Wang, L.; Zhang, Z.; Dong, Z.; Tang, F. Nuclear factor-erythroid 2-related factor 3 (NRF3) is low expressed in colorectal cancer and its down-regulation promotes colorectal cancer malignance through activating EGFR and p38/MAPK. Am. J. Cancer Res. 2019, 9, 511–528. [Google Scholar] [PubMed]
- Chen, L.; Lu, D.; Sun, K.; Xu, Y.; Hu, P.; Li, X.; Xu, F. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene 2019, 692, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, G.G.; Paquet, M.; Blank, V. Nfe2l3 (Nrf3) deficiency predisposes mice to T-cell lymphoblastic lymphoma. Blood 2011, 117, 2005–2008. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB Protein Keap1 Targets Antioxidant Transcription Factor Nrf2 for Ubiquitination by the Cullin 3-Roc1 Ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kobayashi, A.; Yamamoto, M.; Hayes, J.D. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J. Biol. Chem. 2009, 284, 3195–3210. [Google Scholar] [CrossRef] [Green Version]
- Nouhi, Z.; Chevillard, G.G.; Derjuga, A.; Blank, V. Endoplasmic reticulum association and N-linked glycosylation of the human Nrf3 transcription factor. FEBS Lett. 2007, 581, 5401–5406. [Google Scholar] [CrossRef] [Green Version]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yu, H.; Hughes, N.W.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Article Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 2014, 16, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, M.C.J.; Bodnar, N.O.; Rapoport, T.A. Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 2020, 117, 7776–7781. [Google Scholar] [CrossRef] [Green Version]
- Dirac-Svejstrup, A.B.; Walker, J.; Faull, P.; Encheva, V.; Akimov, V.; Puglia, M.; Perkins, D.; Kümper, S.; Hunjan, S.S.; Blagoev, B.; et al. DDI2 Is a Ubiquitin-Directed Endoprotease Responsible for Cleavage of Transcription Factor NRF1. Mol. Cell 2020, 79, 332–341. [Google Scholar] [CrossRef]
- Kannan, M.B.; Dodard-Friedman, I.; Blank, V. Stringent control of NFE2L3 (Nuclear Factor, Erythroid 2-Like 3; NRF3) protein degradation by FBW7 (F-box/WD Repeatcontaining Protein 7) and glycogen synthase kinase 3 (GSK3). J. Biol. Chem. 2015, 290, 26292–26302. [Google Scholar] [CrossRef] [Green Version]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta 2015, 1849, 1260–1276. [Google Scholar] [CrossRef]
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear Factor Erythroid-2 Like 1 (NFE2L1): Structure, function and regulation. Gene 2016, 584, 17–25. [Google Scholar] [CrossRef]
- Chen, L.; Kwong, M.; Lu, R.; Ginzinger, D.; Lee, C.; Leung, L.; Chan, J.Y. Nrf1 Is Critical for Redox Balance and Survival of Liver Cells during Development. Mol. Cell. Biol. 2003, 23, 4673–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Vega, D.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2—Related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 1, 2029–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345. [Google Scholar] [CrossRef] [PubMed]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5, e18357. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5, e17721. [Google Scholar] [CrossRef] [PubMed]
- Sivá, M.; Svoboda, M.; Veverka, V.; Trempe, J.-F.F.; Hofmann, K.; Kožíšek, M.; Hexnerová, R.; Sedlák, F.; Belza, J.; Brynda, J.; et al. Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci. Rep. 2016, 6, 30443. [Google Scholar] [CrossRef] [Green Version]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef]
- Fassmannová, D.; Sedlák, F.; Sedláček, J.; Špička, I.; Šašková, K.G. Nelfinavir inhibits the tcf11/nrf1-mediated proteasome recovery pathway in multiple myeloma. Cancers 2020, 12, 1065. [Google Scholar] [CrossRef]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef] [Green Version]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.X.; Phuektes, P.; Kumar, P.; Goh, G.Y.L.; Moreau, D.; Chow, V.T.K.; Bard, F.; Chu, J.J.H. Human genome-wide RNAi screen reveals host factors required for enterovirus 71 replication. Nat. Commun. 2016, 7, 13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers 2020, 12, 2681. https://doi.org/10.3390/cancers12092681
Kobayashi A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers. 2020; 12(9):2681. https://doi.org/10.3390/cancers12092681
Chicago/Turabian StyleKobayashi, Akira. 2020. "Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors" Cancers 12, no. 9: 2681. https://doi.org/10.3390/cancers12092681
APA StyleKobayashi, A. (2020). Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers, 12(9), 2681. https://doi.org/10.3390/cancers12092681