Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Knock-in of the EGFR C797S Mutation into H1975 Cells
2.2. Knock-in of the EGFR C797S Mutation Produces AZD9291-Resistant Phenotypes
2.3. Exploration of TKI Resistance Mechanisms by Whole-Transcriptome Sequencing and Quantitative Proteomics
2.4. Upregulation of AXL in NSCLC Cell Lines Carrying EGFR C797S
2.5. Effects of AXL Inhibition in NSCLC Cell Lines Carrying EGFR C797S In Vitro
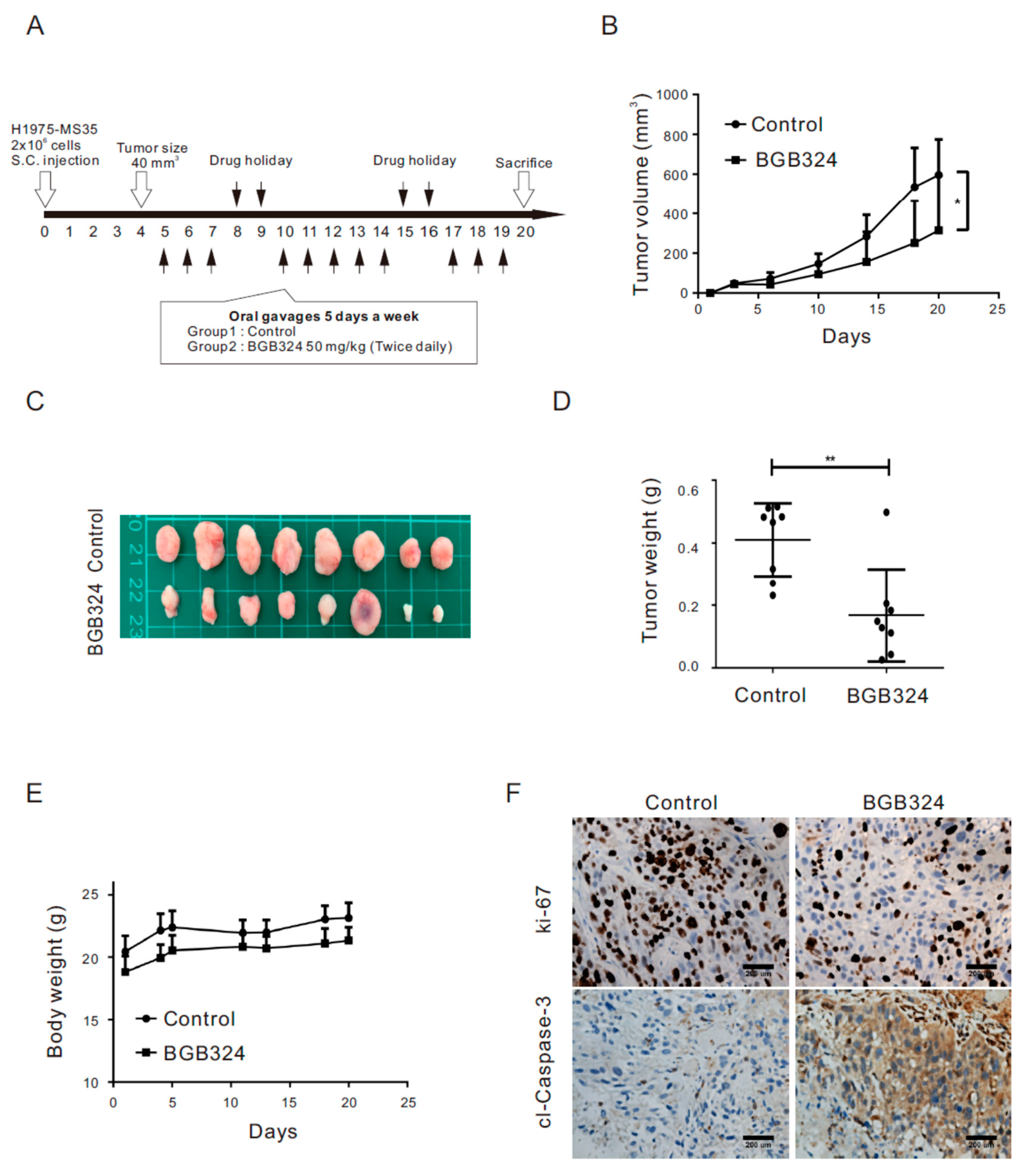
2.6. Inhibition of AXL Represses Tumor Growth in Xenograft Mice Engrafted with H1975 Cells Harboring the EGFR C797S Mutation
3. Discussion
4. Materials and Methods
4.1. Culture Media, Reagents, and Antibodies
4.2. Cell Lines
4.3. Knock-in of the EGFR C797S Mutation by CRISPR/Cas9 Genome Editing
4.4. Detection of EGFR C797S by PCR
4.5. Sanger Sequence Analysis
4.6. Generation of the AZD9291-Resistant H1975-R Cell Line
4.7. Plasmids, Transfection, and Lentivirus Production and Infection
4.8. Proteome Analysis with Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Mass Spectrometry (MS)
4.9. Pathway and Network Analyses
4.10. Whole-Transcriptome Sequencing
4.11. siRNA and Transfection
4.12. Cell Viability Assays
4.13. Assay for Colony Forming Ability
4.14. Western Blot
4.15. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
4.16. Xenograft Mouse Tumor Model
4.17. Immunohistochemistry (IHC)
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gower, A.; Wang, Y.; Giaccone, G. Oncogenic drivers, targeted therapies, and acquired resistance in non-small-cell lung cancer. J. Mol. Med. 2014, 92, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardou, H.; Dahabreh, I.J.; Bafaloukos, D.; Kosmidis, P.; Murray, S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat. Rev. Clin. Oncol. 2009, 6, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-S.; Kumarakulasinghe, N.B.; Huang, Y.-Q.; Ang, Y.L.E.; Choo, J.R.-E.; Goh, B.-C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Landi, L.; Cappuzzo, F. Irreversible EGFR-TKIs: Dreaming perfection. Transl. Lung Cancer Res. 2013, 2, 40–49. [Google Scholar]
- Mazza, V.; Cappuzzo, F. Treating EGFR mutation resistance in non-small cell lung cancer—Role of osimertinib. Appl. Clin. Genet. 2017, 10, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patients with Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nat. Cell Biol. 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. New Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.-H.; Lu, J.-J. Osimertinib resistance in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Lett. 2018, 420, 242–246. [Google Scholar] [CrossRef]
- Ren, X.; Cai, X.; Li, J.; Zhang, X.; Yu, J.; Song, X.; Zhang, H.; Song, X. Histological transformation of lung adenocarcinoma to small cell lung cancer with mutant C797S conferring acquired resistance to osimertinib. J. Int. Med Res. 2020, 48, 0300060520927918. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Warner, S.L.; Vankayalapati, H.; Bearss, D.J.; Sharma, S. Targeting Axl and Mer Kinases in Cancer. Mol. Cancer Ther. 2011, 10, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.-Y.; Stehr, H.; Von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tsai, M.; Wu, S.; Chang, T.; Tsai, T.; Gow, C.; Chang, Y.; Shih, J.-Y. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int. J. Cancer 2019, 145, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL Mediates Resistance to Cetuximab Therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Yang, G.; Feng, M.; Cao, Z.; Liu, Y.; Qiu, J.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. Expression, function and clinical application of stanniocalcin-1 in cancer. J. Cell Mol. Med. 2020, 24, 7686–7696. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Y.-J.; Man, Y.; Pan, F.; Li, Z.-H.; Jia, L.-S. Knockdown of AXL Receptor Tyrosine Kinase in Osteosarcoma Cells Leads to Decreased Proliferation and Increased Apoptosis. Int. J. Immunopathol. Pharmacol. 2013, 26, 179–188. [Google Scholar] [CrossRef]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Barón, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Song, Q.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466–1482. [Google Scholar] [PubMed]
- Chen, C.-H.; Changou, C.A.; Hsieh, T.-H.; Lee, Y.-C.; Chu, C.-Y.; Hsu, K.-C.; Wang, H.-C.; Lin, Y.-C.; Lo, Y.-N.; Liu, Y.-R.; et al. Dual Inhibition of PIK3C3 and FGFR as a New Therapeutic Approach to Treat Bladder Cancer. Clin. Cancer Res. 2017, 24, 1176–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.-Y.; Kao, S.-H.; Wang, W.-L.; Chen, C.-Y.; Hsiao, T.-H.; Salunke, S.B.; Chen, J.J.W.; Su, K.-Y.; Yang, S.-C.; Hong, T.-M.; et al. Small Molecule T315 Promotes Casitas B-Lineage Lymphoma–Dependent Degradation of Epidermal Growth Factor Receptor via Y1045 Autophosphorylation. Am. J. Respir. Crit. Care Med. 2016, 193, 753–766. [Google Scholar] [CrossRef]
- Lee, K.-M.; Wu, C.-C.; Wu, S.-E.; Lin, Y.-H.; Wang, L.-T.; Chang, C.-R.; Huang, P.-N.; Shih, S.-R.; Chien-Kuo, L. The RNA-dependent RNA polymerase of enterovirus A71 associates with ribosomal proteins and positively regulates protein translation. RNA Biol. 2020, 17, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Wu, C.-C.; Huang, K.-Y.; Chuang, W.-Y.; Hsueh, C.; Li, H.-J.; Chen, C.-Y. Profiling of subcellular EGFR interactome reveals hnRNP A3 modulates nuclear EGFR localization. Oncogenesis 2020, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, C.-C.; Chuang, W.-Y.; Leu, Y.-L.; Ueng, S.-H.; Hsueh, C.; Yeh, C.-T.; Wang, T.-H. Hydroxygenkwanin Inhibits Class I HDAC Expression and Synergistically Enhances the Antitumor Activity of Sorafenib in Liver Cancer Cells. Front. Oncol. 2020, 10, 216. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Chen, C.-Y.; Ueng, S.-H.; Hsueh, C.; Yeh, C.-T.; Ho, J.-Y.; Chou, L.-F.; Wang, T.-H. Corylin increases the sensitivity of hepatocellular carcinoma cells to chemotherapy through long noncoding RNA RAD51-AS1-mediated inhibition of DNA repair. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Jan, C.-I.; Lo, J.-F.; Yang, S.-C.; Chang, Y.-L.; Pan, S.-H.; Wang, W.-L.; Hong, T.-M.; Yang, P.-C. Tid1-L Inhibits EGFR Signaling in Lung Adenocarcinoma by Enhancing EGFR Ubiquitinylation and Degradation. Cancer Res. 2013, 73, 4009–4019. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Jan, C.-I.; Pi, W.-C.; Wang, W.-L.; Yang, P.-C.; Wang, T.-H.; Karni, R.; Wang, T.-C.V. Heterogeneous nuclear ribonucleoproteins A1 and A2 modulate expression of Tid1 isoforms and EGFR signaling in non-small cell lung cancer. Oncotarget 2016, 7, 16760–16772. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process | p Value a | Identified Proteins Involved in the Process |
|---|---|---|
| myosin heavy chain binding | 0.0000 | LIMCH1, CORO1A, AXL |
| extracellular matrix structural constituent conferring tensile strength | 0.0001 | COL17A1, COL7A1, COL21A1, COL4A5 |
| extracellular matrix structural constituent | 0.0002 | COL17A1, COL7A1, COL21A1, HSPG2, TINAGL1, COL4A5 |
| myosin binding | 0.0006 | LIMCH1, CORO1A, LMTK2, AXL |
| actin binding | 0.0006 | SCIN, LIMCH1, EPB41L2, CORO1A, SYNE1, KCNMA1, PKNOX2, DNASE1, ANXA8L1 |
| 1-phosphatidylinositol binding | 0.0031 | SCIN, FRMPD2 |
| cysteine-type endopeptidase activity involved in apoptotic process | 0.0031 | CASP14,CASP1 |
| endodeoxyribonuclease activity, producing 5’-phosphomonoesters | 0.0035 | DNASE1L3, DNASE1 |
| actin filament binding | 0.0042 | SCIN, CORO1A, SYNE1, PKNOX2, ANXA8L1 |
| serine-type peptidase activity | 0.0042 | F3, DPP6, TMPRSS4, MMP13, KLK6, DPP4 |
| serine hydrolase activity | 0.0045 | F3, DPP6, TMPRSS4, MMP13, KLK6, DPP4 |
| protease binding | 0.0052 | TIMP2, SERPINE1, F3, DPP4 |
| 3’,5’-cyclic-GMP phosphodiesterase activity | 0.0054 | PDE1C, PDE2A |
| virus receptor activity | 0.0081 | NECTIN1, AXL, DPP4 |
| hijacked molecular function | 0.0081 | NECTIN1, AXL, DPP4 |
| chloride channel activity | 0.0084 | FXYD3, SLC26A7, GABRB3 |
| actin monomer binding | 0.0098 | CORO1A, PKNOX2 |
| anion transmembrane transporter activity | 0.0103 | FXYD3, SLC6A12, SLC37A2, SLC26A7, GABRB3, SLCO2A1 |
| 3’,5’-cyclic-nucleotide phosphodiesterase activity | 0.0105 | PDE1C, PDE2A |
| cyclic-nucleotide phosphodiesterase activity | 0.0113 | PDE1C, PDE2A |
| Biological Process a | Number of Identified Proteins Involved in the Process | False Discovery Rate |
|---|---|---|
| extracellular matrix organization | 9 | 0.0028 |
| cell junction organization | 7 | 0.0074 |
| cell junction assembly | 6 | 0.0075 |
| multicellular organismal process | 43 b | 0.0087 |
| cellular response to extracellular stimulus | 7 b | 0.0110 |
| anatomical structure development | 36 b | 0.0121 |
| anatomical structure formation involved in morphogenesis | 12 | 0.0170 |
| regulation of cellular component movement | 12 | 0.0261 |
| multicellular organism development | 33 b | 0.0273 |
| response to external stimulus | 18 b | 0.0273 |
| cellular component organization | 35 b | 0.0273 |
| cell adhesion | 11 b | 0.0393 |
| tissue development | 16 | 0.0394 |
| cell-substrate adhesion | 5 b | 0.0394 |
| regulation of localization | 21 b | 0.0394 |
| anatomical structure morphogenesis | 18 b | 0.0398 |
| system development | 29 b | 0.0406 |
| regulation of locomotion | 11 | 0.0422 |
| positive regulation of cellular component movement | 8 | 0.0422 |
| regulation of cell adhesion | 9 | 0.0427 |
| regulation of cell migration | 10 | 0.0427 |
| regulation of biological quality | 26 b | 0.0427 |
| positive regulation of locomotion | 8 | 0.0450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.-H.; Wu, C.-C.; Huang, K.-Y.; Leu, Y.-L.; Yang, S.-C.; Chen, C.-L.; Chen, C.-Y. Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer. Cancers 2021, 13, 111. https://doi.org/10.3390/cancers13010111
Wang T-H, Wu C-C, Huang K-Y, Leu Y-L, Yang S-C, Chen C-L, Chen C-Y. Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer. Cancers. 2021; 13(1):111. https://doi.org/10.3390/cancers13010111
Chicago/Turabian StyleWang, Tong-Hong, Chih-Ching Wu, Kuo-Yen Huang, Yann-Lii Leu, Shuenn-Chen Yang, Ci-Ling Chen, and Chi-Yuan Chen. 2021. "Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer" Cancers 13, no. 1: 111. https://doi.org/10.3390/cancers13010111