Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
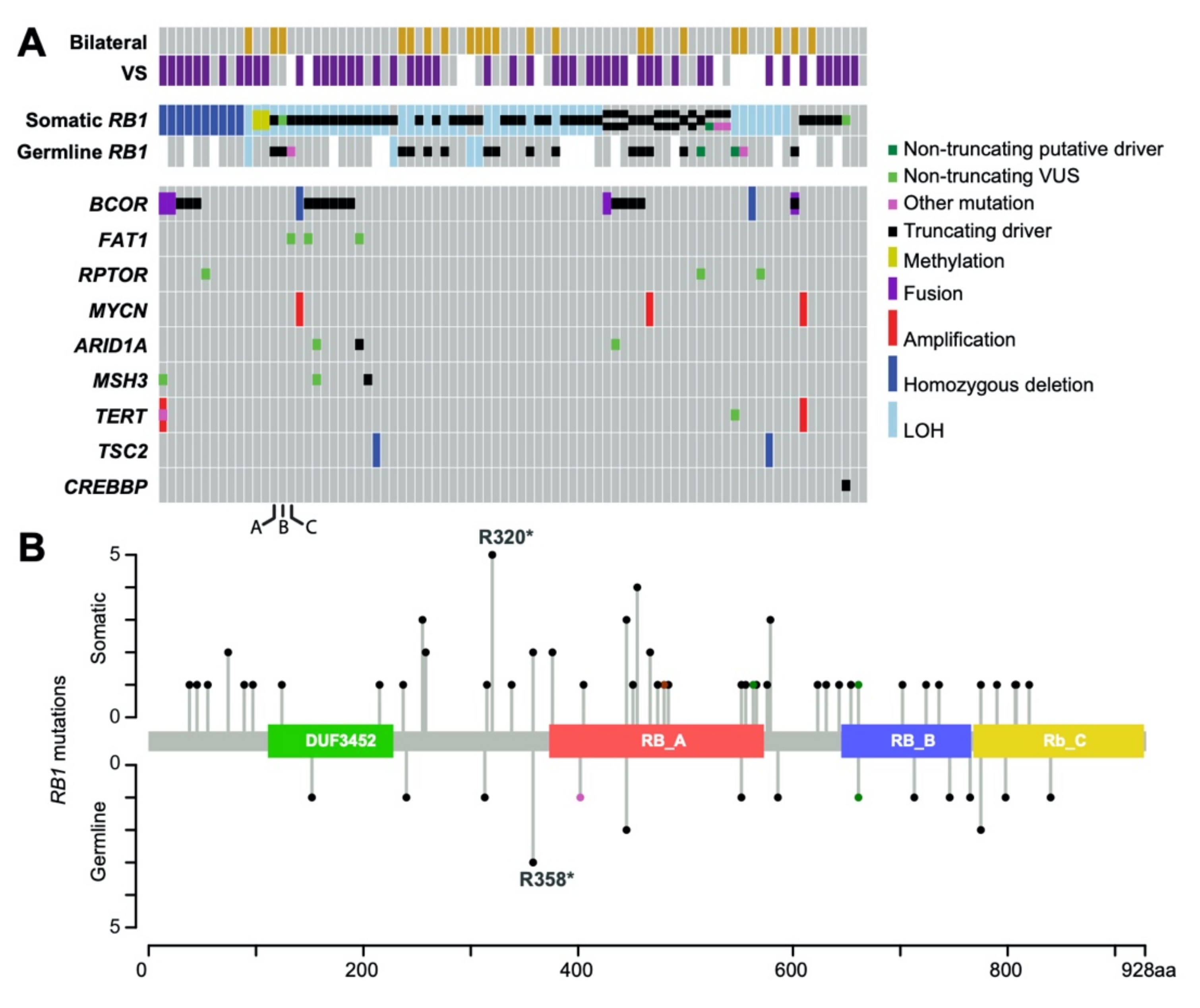
2.1. Genomic Landscape of RB1 Alterations in Retinoblastoma
2.2. Evidence of Intertumoral Genetic Heterogeneity
2.3. Non-RB1 Alterations in Retinoblastoma
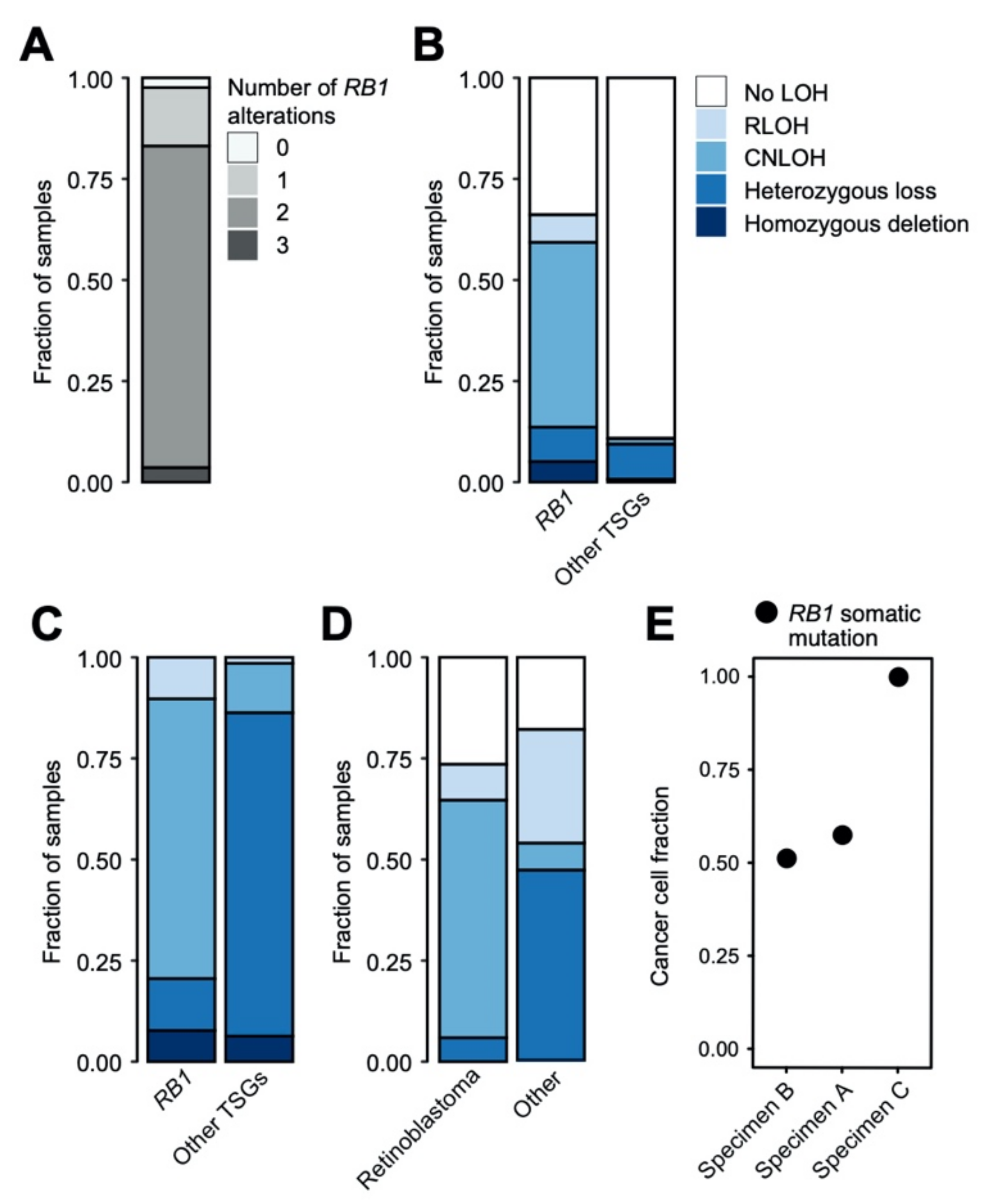
2.4. Copy Number Alterations and Loss of Heterozygosity in Retinoblastoma
3. Discussion
4. Materials and Methods
4.1. Isolation and Purification of DNA
4.2. Exon-Capture Sequencing
4.3. Outside Testing of RB1 Alterations
4.4. Biostatistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abramson, D.H. Retinoblastoma: Saving life with vision. Annu. Rev. Med. 2014, 65, 171–184. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Brandt, B.; Höpping, W.; Passarge, E.; Horsthemke, B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum. Genet. 1994, 94, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corson, T.W.; Gallie, B.L. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosom. Cancer 2007, 46, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Gratias, S.; Rieder, H.; Ullmann, R.; Klein-Hitpass, L.; Schneider, S.; Bölöni, R.; Kappler, M.; Lohmann, D.R. Allelic Loss in a Minimal Region on Chromosome 16q24 Is Associated with Vitreous Seeding of Retinoblastoma. Cancer Res. 2007, 67, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Herzog, S.; Lohmann, D.; Buiting, K.; Schüler, A.; Horsthemke, B.; Rehder, H.; Rieder, H. Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization. Hum. Genet. 2001, 108, 98–104. [Google Scholar] [CrossRef]
- Kooi, I.E.; Mol, B.M.; Massink, M.P.G.; Ameziane, N.; Meijers-Heijboer, H.; Dommering, C.J.; van Mil, S.E.; de Vries, Y.; van der Hout, A.H.; Kaspers, G.J.L.; et al. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci. Rep. 2016, 6, 25264. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, J.; Nagahawatte, P.; Finkelstein, D.; Richards-Yutz, J.; Valentine, M.; Ma, J.; Mullighan, C.; Song, G.; Chen, X.; Wilson, M.; et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 2014, 5, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Murphree, A.L.; Benedict, W.F. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef]
- Ohtani-Fujita, N.; Dryja, T.P.; Rapaport, J.M.; Fujita, T.; Matsumura, S.; Ozasa, K.; Watanabe, Y.; Hayashi, K.; Maeda, K.; Kinoshita, S.; et al. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet. Cytogenet. 1997, 98, 43–49. [Google Scholar] [CrossRef]
- Mallipatna, A.; Marino, M.; Singh, A.D. Genetics of Retinoblastoma. Asia Pac. J. Ophthal. 2016, 5, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Bowles, E.; Corson, T.W.; Bayani, J.; Squire, J.A.; Wong, N.; Lai, P.B.-S.; Gallie, B.L. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosom. Cancer 2007, 46, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Marchong, M.N.; Chen, D.; Corson, T.W.; Lee, C.; Harmandayan, M.; Bowles, E.; Chen, N.; Gallie, B.L. Minimal 16q genomic loss implicates cadherin-11 in retinoblastoma. Mol. Cancer Res. 2004, 2, 495–503. [Google Scholar]
- Van der Wal, J.E.; Hermsen, M.A.J.A.; Gille, H.J.P.; Schouten-Van Meeteren, N.Y.N.; Moll, A.C.; Imhof, S.M.; Meijer, G.A.; Baak, J.P.A.; van der Valk, P. Comparative genomic hybridisation divides retinoblastomas into a high and a low level chromosomal instability group. J. Clin. Pathol. 2003, 56, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, D.-L.; Cobrinik, D. MDM2 but not MDM4 promotes retinoblastoma cell proliferation through p53-independent regulation of MYCN translation. Oncogene 2017, 36, 1760–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef]
- Grotta, S.; D’Elia, G.; Scavelli, R.; Genovese, S.; Surace, C.; Sirleto, P.; Cozza, R.; Romanzo, A.; De Ioris, M.A.; Valente, P. Advantages of a next generation sequencing targeted approach for the molecular diagnosis of retinoblastoma. BMC Cancer 2015, 15, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amitrano, S.; Marozza, A.; Somma, S.; Imperatore, V.; Hadjistilianou, T.; De Francesco, S.; Toti, P.; Galimberti, D.; Meloni, I.; Cetta, F. Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur. J. Hum. Genet. 2015, 23, 1523–1530. [Google Scholar] [CrossRef]
- Devarajan, B.; Prakash, L.; Kannan, T.R.; Abraham, A.A.; Kim, U.; Muthukkaruppan, V.; Vanniarajan, A. Targeted next generation sequencing of RB1 gene for the molecular diagnosis of Retinoblastoma. BMC Cancer 2015, 15, 320. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scot, N.S. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Ahani, A.; Behnam, B.; Khorshid, H.R.K.; Akbari, M.T. RB1 gene mutations in Iranian patients with retinoblastoma: Report of four novel mutations. Cancer Genet. 2011, 204, 316–322. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Lillington, D.M.; Kingston, J.E.; Coen, P.G.; Price, E.; Hungerford, J.; Domizio, P.; Young, B.D.; Onadim, Z. Comparative genomic hybridization of 49 primary retinoblastoma tumors identifies chromosomal regions associated with histopathology, progression, and patient outcome. Genes Chromosom. Cancer 2002, 36, 121–128. [Google Scholar] [CrossRef]
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; Van Ziffle, J.; Onodera, C. Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 2020, 127, 804–813. [Google Scholar] [CrossRef]
- Gustmann, S.; Klein-Hitpass, L.; Stephan, H.; Weber, S.; Bornfeld, N.; Kaulisch, M.; Lohmann, D.R.; Dünker, N. Loss at chromosome arm 16q in retinoblastoma: Confirmation of the association with diffuse vitreous seeding and refinement of the recurrently deleted region. Genes Chromosom. Cancer 2011, 50, 327–337. [Google Scholar] [CrossRef]
- Xu, L.; Polski, A.; Prabakar, R.K.; Reid, M.W.; Chévez-Barrios, P.; Jubran, R.; Kim, J.W.; Kuhn, P.; Cobrinik, D.; Hicks, J.; et al. Chromosome 6p amplification in aqueous humor cell-free DNA is a prognostic biomarker for retinoblastoma ocular survival. Mol. Cancer Res. 2020, 18, 1166–1675. [Google Scholar] [CrossRef]
- Berry, J.L.; Xu, L.; Kooi, I.; Murphree, A.L.; Prabakar, R.K.; Reid, M.; Stachelek, K.; Le, B.H.A.; Welter, L.; Reiser, B.J.; et al. Genomic cfDNA Analysis of Aqueous Humor in Retinoblastoma Predicts Eye Salvage: The Surrogate Tumor Biopsy for Retinoblastoma. Mol. Cancer Res. 2018, 16, 1701–1712. [Google Scholar] [CrossRef] [Green Version]
- Andersen, C.L.; Wiuf, C.; Kruhøffer, M.; Korsgaard, M.; Laurberg, S.; Ørntoft, T.F. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis 2007, 28, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, C.; McDevitt, M.A.; Maciejewski, J.P. Copy neutral loss of heterozygosity: A novel chromosomal lesion in myeloid malignancies. Blood 2010, 115, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, M.; Smith, L.-L.; Lillington, D.M.; Chaplin, T.; Kakkas, I.; Molloy, G.; Chelala, C.; Cazier, J.-B.; Cavenagh, J.D.; Fitzgibbon, J.; et al. Segmental uniparental disomy is a commonly acquired genetic event in relapsed acute myeloid leukemia. Blood 2008, 112, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, S.K.; DiFrancesco, L.M.; Ogilvie, R.T.; Demetrick, D.J. Loss of heterozygosity associated with uniparental disomy in breast carcinoma. Mod. Pathol. 2002, 15, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Torabi, K.; Miró, R.; Fernández-Jiménez, N.; Quintanilla, I.; Ramos, L.; Prat, E.; del Rey, J.; Pujol, N.; Killian, J.K.; Meltzer, P.; et al. Patterns of somatic uniparental disomy identify novel tumor suppressor genes in colorectal cancer. Carcinogenesis 2015, 36, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astolfi, A.; Fiore, M.; Melchionda, F.; Indio, V.; Bertuccio, S.N.; Pession, A. BCOR involvement in cancer. Epigenomics 2019, 11, 835–855. [Google Scholar] [CrossRef] [Green Version]
- Terada, K.; Yamaguchi, H.; Ueki, T.; Usuki, K.; Kobayashi, Y.; Tajika, K.; Gomi, S.; Kurosawa, S.; Saito, R.; Furuta, Y.; et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosom. Cancer 2018, 57, 401–408. [Google Scholar] [CrossRef]
- Zhou, Y.; Wojcik, A.; Sanders, V.R.; Rahmani, B.; Kurup, S.P. Ocular findings in a patient with oculofaciocardiodental (OFCD) syndrome and a novel BCOR pathogenic variant. Int. Ophthalmol. 2018, 38, 2677–2682. [Google Scholar] [CrossRef]
- Zeng, Y.; He, T.; Liu, J.; Li, Z.; Xie, F.; Chen, C.; Xing, Y. Bioinformatics analysis of multi-omics data identifying molecular biomarker candidates and epigenetically regulatory targets associated with retinoblastoma. Medicine 2020, 99, e23314. [Google Scholar] [CrossRef]
- Yazici, H.; Wu, H.-C.; Tigli, H.; Yilmaz, E.Z.; Kebudi, R.; Santella, R.M. High levels of global genome methylation in patients with retinoblastoma. Oncol. Lett. 2020, 20, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs. Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Wang, Z.; Liu, Q.; Song, N.; Wilson, C.L.; Ehrhardt, M.J.; Shelton, K.; Easton, J.; Mulder, H.; Kennetz, D. Pathogenic Germline Mutations in DNA Repair Genes in Combination With Cancer Treatment Exposures and Risk of Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J. Clin. Oncol. 2020, 38, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhai, Y.; Shi, R.; Qian, Y.; Cui, H.; Yang, J.; Bi, Y.; Yan, T.; Yang, J.; Ma, Y. FAT1 inhibits cell migration and invasion by affecting cellular mechanical properties in esophageal squamous cell carcinoma. Oncol. Rep. 2018, 39, 2136–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.-C. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients with Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- De Toni, L.; Šabovic, I.; Cosci, I.; Ghezzi, M.; Foresta, C.; Garolla, A. Testicular Cancer: Genes, Environment, Hormones. Front. Endocrinol. 2019, 10, 408. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, J.H.; Richards, A.L.; Mandelker, D.L.; Berger, M.F.; Walsh, M.F.; Dunkel, I.J.; Donoghue, M.T.A.; Abramson, D.H. Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers 2021, 13, 149. https://doi.org/10.3390/cancers13010149
Francis JH, Richards AL, Mandelker DL, Berger MF, Walsh MF, Dunkel IJ, Donoghue MTA, Abramson DH. Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers. 2021; 13(1):149. https://doi.org/10.3390/cancers13010149
Chicago/Turabian StyleFrancis, Jasmine H., Allison L. Richards, Diana L. Mandelker, Michael F. Berger, Michael F. Walsh, Ira J. Dunkel, Mark T. A. Donoghue, and David H. Abramson. 2021. "Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing" Cancers 13, no. 1: 149. https://doi.org/10.3390/cancers13010149
APA StyleFrancis, J. H., Richards, A. L., Mandelker, D. L., Berger, M. F., Walsh, M. F., Dunkel, I. J., Donoghue, M. T. A., & Abramson, D. H. (2021). Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers, 13(1), 149. https://doi.org/10.3390/cancers13010149