Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
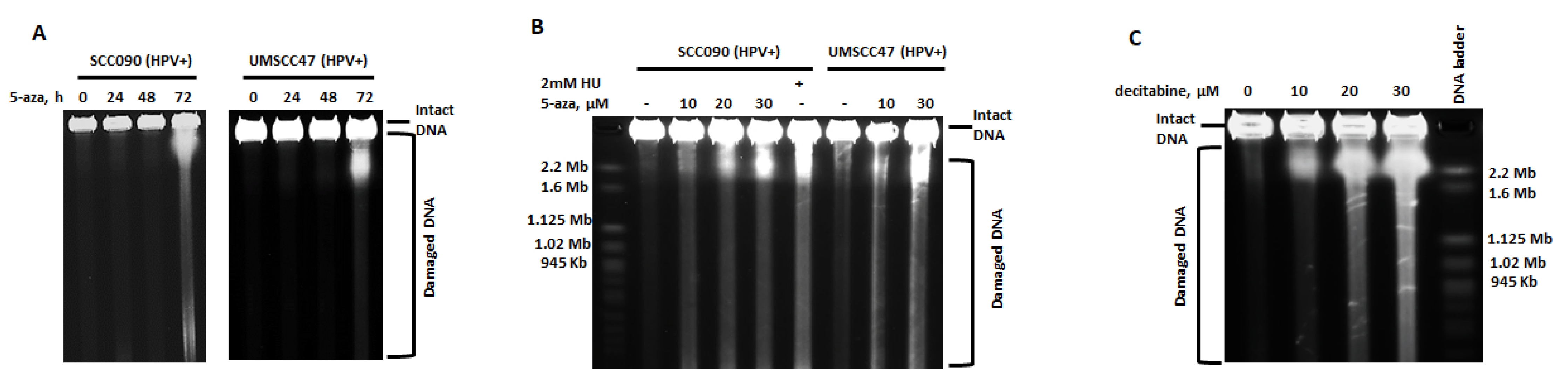
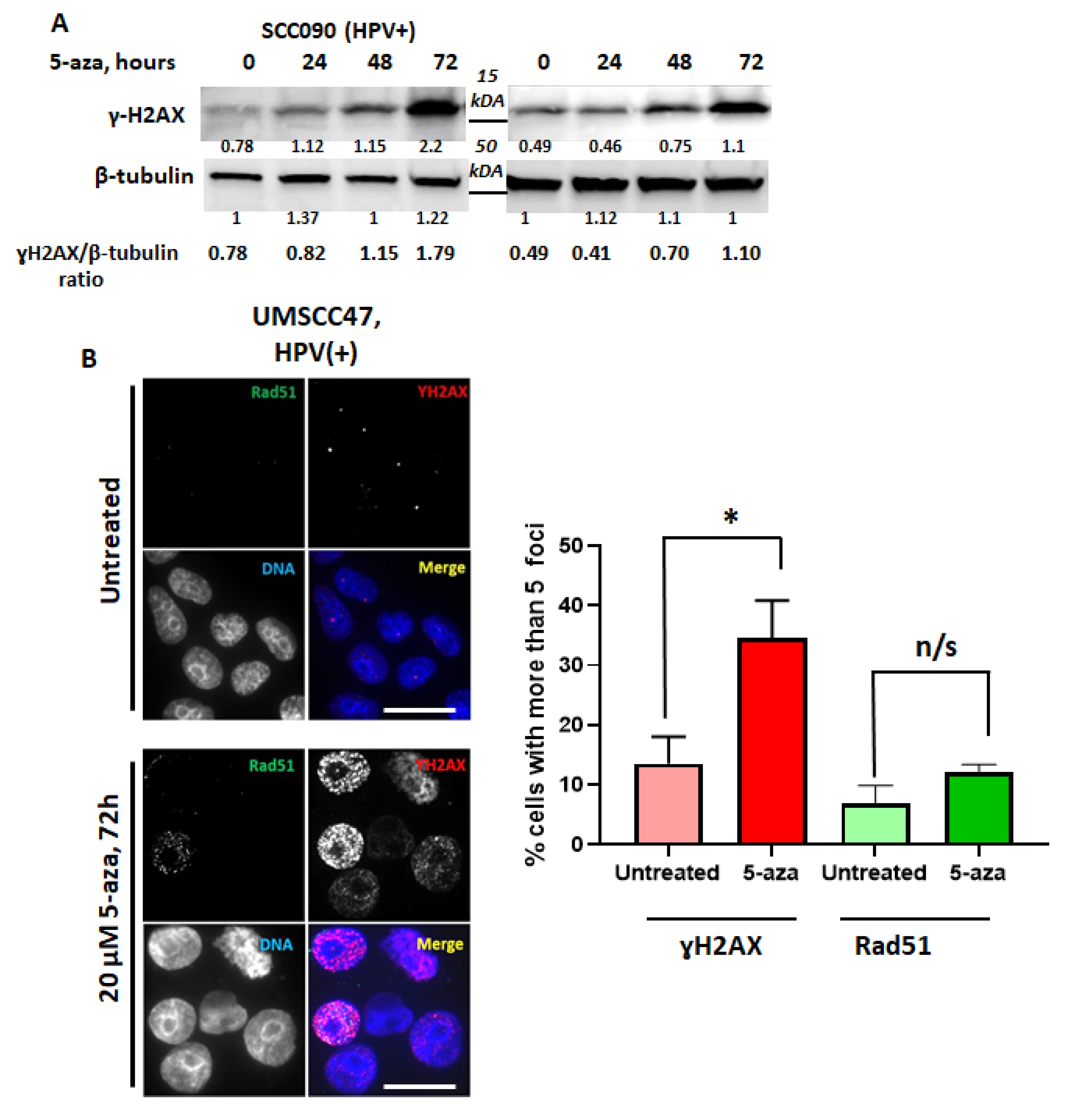
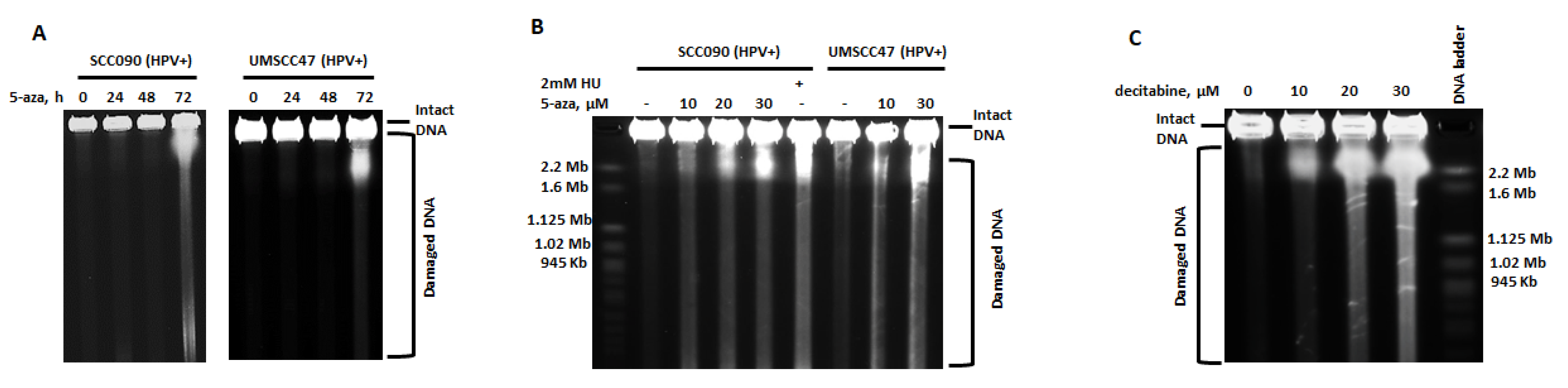
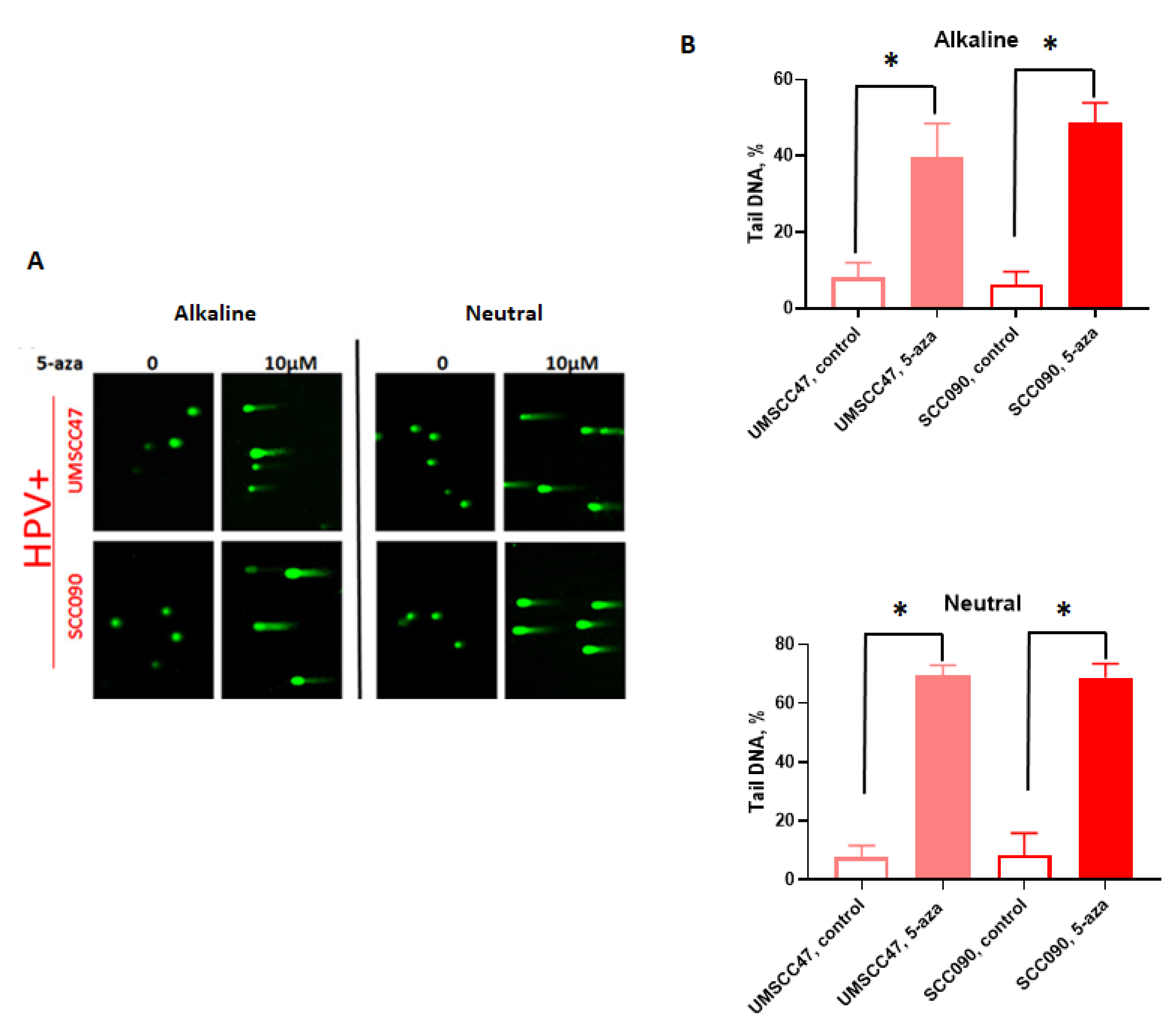
2.1. 5-Azacytidine Causes DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells
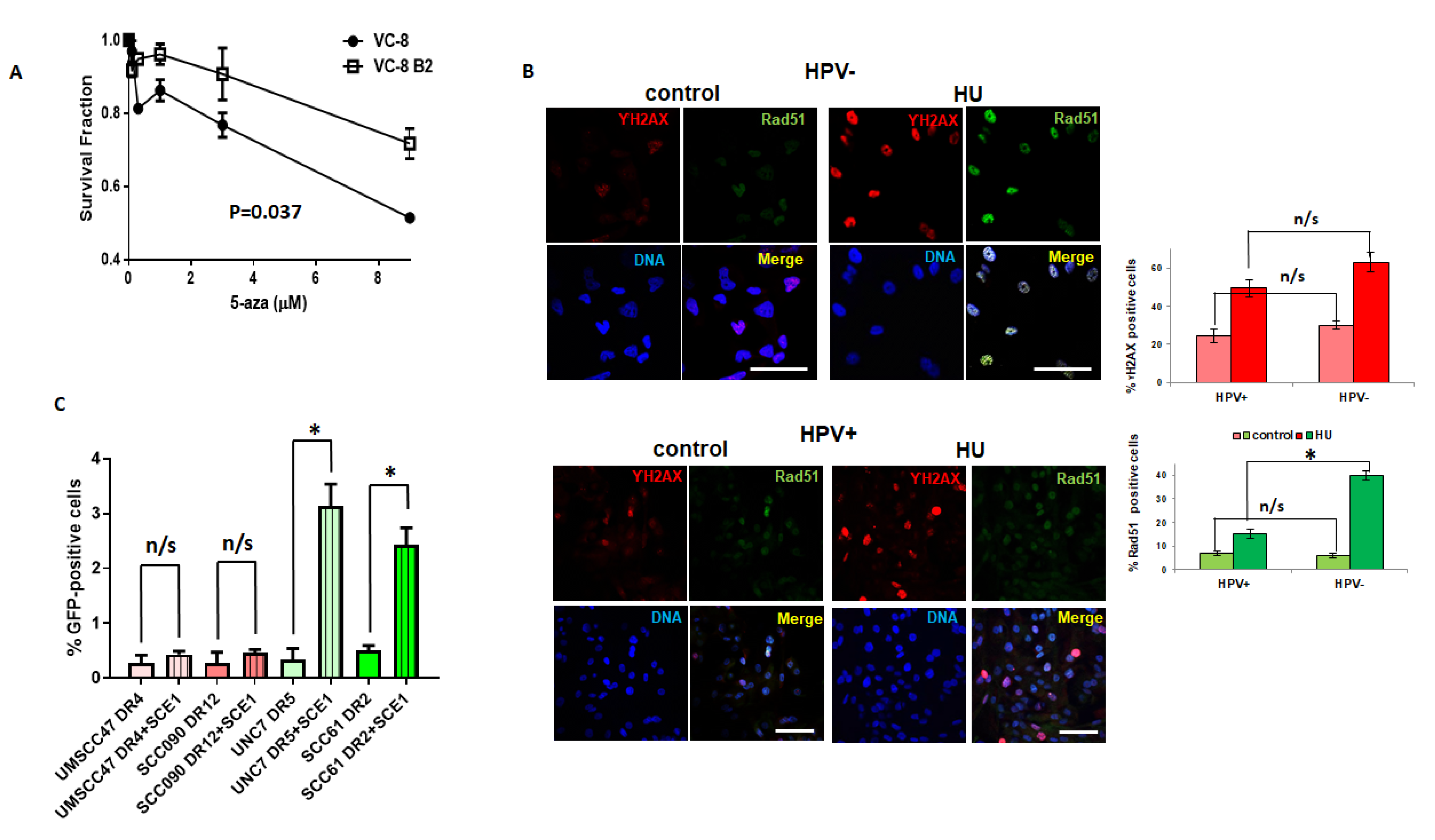
2.2. Dysfunctional Homologous Recombination Increases Sensitivity to 5-azaC
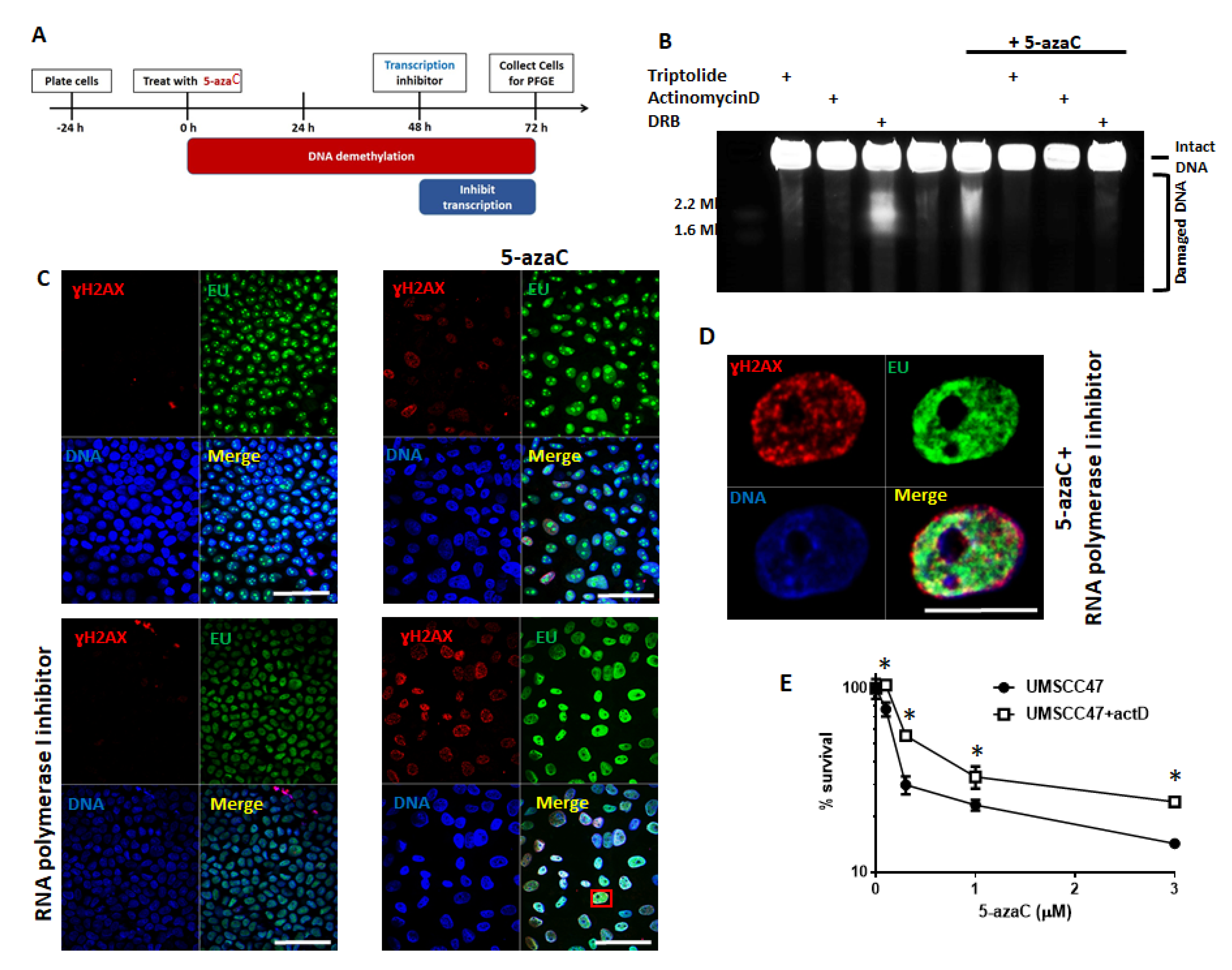
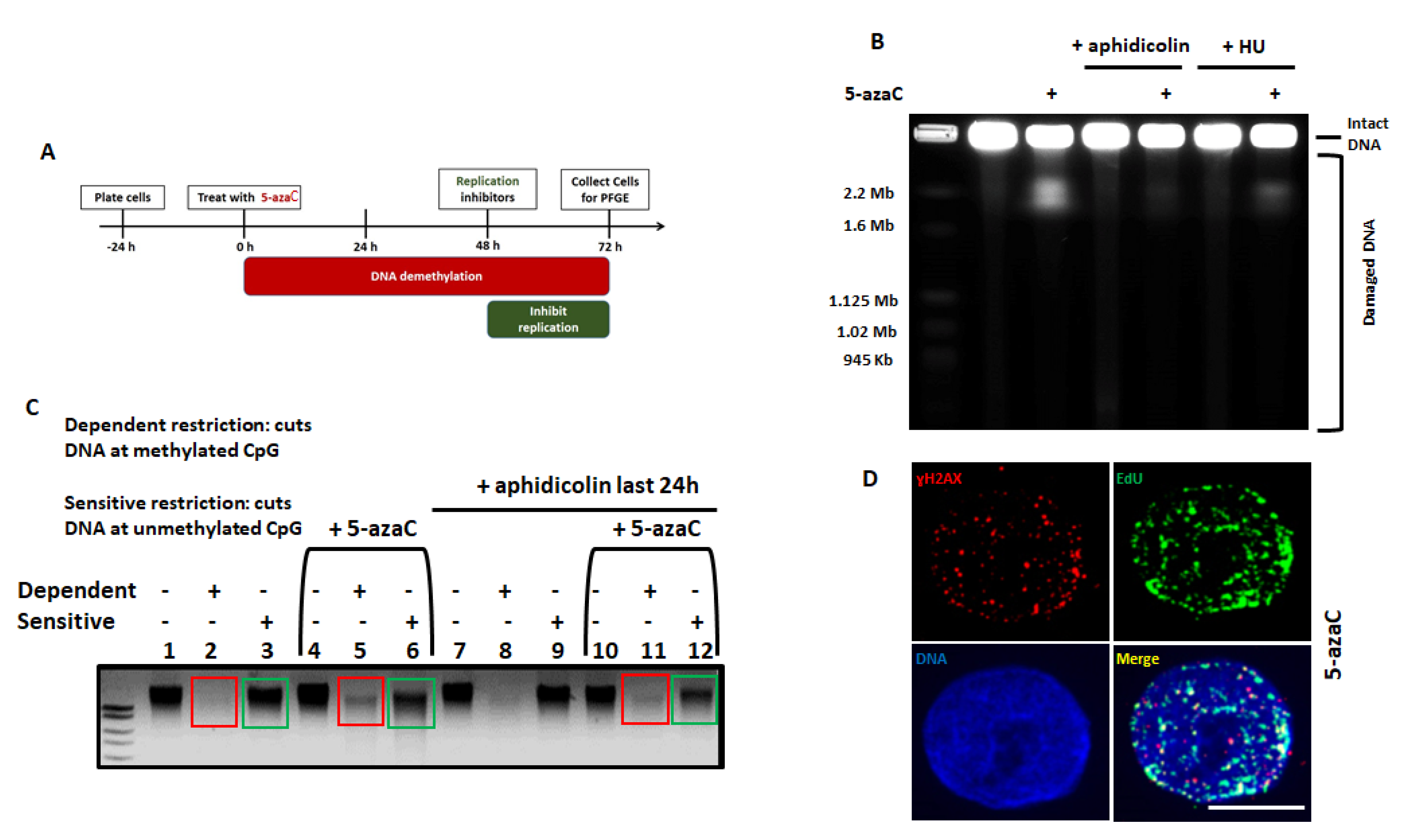
2.3. Transcription and Replication are Required for 5-Azycytidine Induced DNA Double Strand Breaks
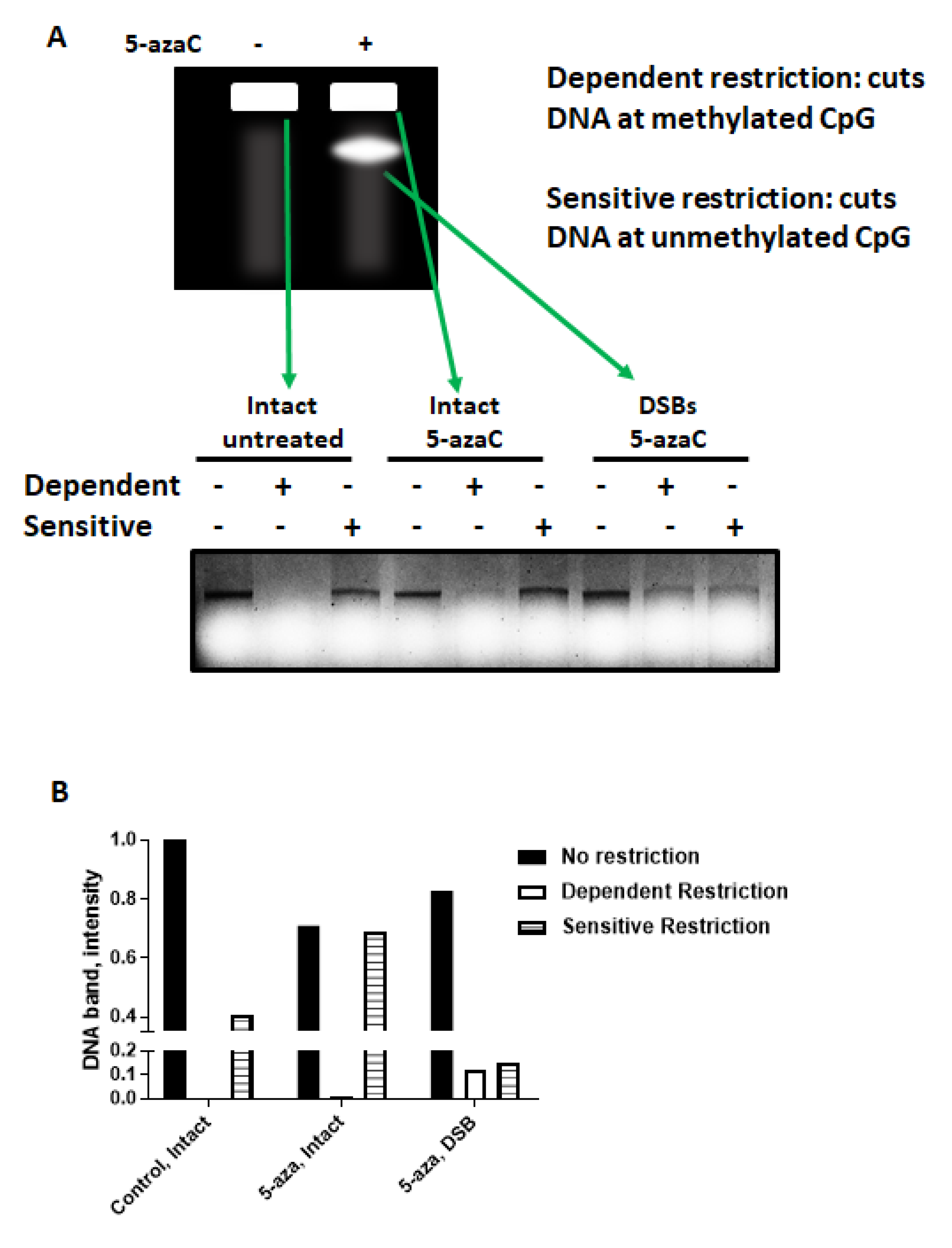
2.4. 5-azaC-Induced DNA DSBs Are Randomly Distributed in the Genome but Are Enriched in Demethylated DNA
2.5. Demethylation Alters the Protein Complement Associated with Chromatin in HPV+ HNSCC
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Transfection
4.3. Drugs
4.4. Homologous Recombination Reporter
4.5. Immunoblotting
4.6. Pulsed-Field Gel Electrophoresis
4.7. Immunofluorescence
4.8. Colony Formation Assay
4.9. DNA Purification and Restriction
4.10. Subcellular Fractionation
4.11. Mass Spectrometry
4.12. Next Generation Sequencing and Analysis
4.13. Comet Assay
4.14. GSEA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2016. [Google Scholar] [CrossRef]
- Gillison, M.L. Current topics in the epidemiology of oral cavity and oropharyngeal cancers. Head Neck 2007, 29, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.K.; Stanley, M.A.; Ebmeyer, J.; Steinstrasser, L.; Upile, T.; Jerjes, W.; Bernal-Sprekelsen, M.; Gorner, M.; Sudhoff, H.H. HPV & head and neck cancer: A descriptive update. Head Neck Oncol. 2009, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Issaeva, N.; Yarbrough, W.G. HPV-driven oropharyngeal cancer: Current knowledge of molecular biology and mechanisms of carcinogenesis. Cancers Head Neck 2018, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forastiere, A.A.; Zhang, Q.; Weber, R.S.; Maor, M.H.; Goepfert, H.; Pajak, T.F.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; et al. Long-Term Results of RTOG 91-11: A Comparison of Three Nonsurgical Treatment Strategies to Preserve the Larynx in Patients With Locally Advanced Larynx Cancer. J. Clin. Oncol. 2013, 31, 845–852. [Google Scholar] [CrossRef]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [Green Version]
- Cannataro, V.L.; Gaffney, S.G.; Sasaki, T.; Issaeva, N.; Grewal, N.K.S.; Grandis, J.R.; Yarbrough, W.G.; Burtness, B.; Anderson, K.S.; Townsend, J.P. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Yarbrough, W.G.; Issaeva, N. Advances in biomarkers and treatment strategies for HPV-associated head and neck cancer. Oncoscience 2018, 5, 140–141. [Google Scholar] [CrossRef]
- Sewell, A.; Brown, B.; Biktasova, A.; Mills, G.B.; Lu, Y.; Tyson, D.R.; Issaeva, N.; Yarbrough, W.G. Reverse-Phase Protein Array Profiling of Oropharyngeal Cancer and Significance of PIK3CA Mutations in HPV-Associated Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- Slebos, R.J.; Yi, Y.; Ely, K.; Carter, J.; Evjen, A.; Zhang, X.; Shyr, Y.; Murphy, B.M.; Cmelak, A.J.; Burkey, B.B.; et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slebos, R.J.; Jehmlich, N.; Brown, B.; Yin, Z.; Chung, C.H.; Yarbrough, W.G.; Liebler, D.C. Proteomic analysis of oropharyngeal carcinomas reveals novel HPV-associated biological pathways. Int. J. Cancer 2013, 132, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 mutations by HPV status in head and neck cancer: Differences in survival and response to DNA-damaging agents. Cancers Head Neck 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer J. Int. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009, 113, 1315–1325. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Diab-Assef, M.; Rohrbeck, A.; Foltzer-Jourdainne, C.; Boltze, C.; Hartig, R.; Schonfeld, P.; Roessner, A.; Gali-Muhtasib, H. 5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3a and induces apoptosis in HCT-116 colon cancer cells via Gadd45- and p53-dependent mechanisms. J. Pharmaco Exp. Ther. 2005, 312, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Katafuchi, A.; Matsubara, M.; Terato, H.; Tsuboi, T.; Masuda, T.; Tatsumoto, T.; Pack, S.P.; Makino, K.; Croteau, D.L.; et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009, 284, 27065–27076. [Google Scholar] [CrossRef] [Green Version]
- Orta, M.L.; Calderon-Montano, J.M.; Dominguez, I.; Pastor, N.; Burgos-Moron, E.; Lopez-Lazaro, M.; Cortes, F.; Mateos, S.; Helleday, T. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Biktasova, A.K.; Hajek, M.; Sewell, A.B.; Gary, C.S.; Bellinger, G.; Deshpande, H.; Bhatia, A.K.; Burtness, B.A.; Judson, B.L.; Mehra, S.; et al. Demethylation therapy as a targeted treatment for human papilloma virus-associated head and neck cancer. Clinical. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Dok, R.; Kalev, P.; Van Limbergen, E.J.; Asbagh, L.A.; Vazquez, I.; Hauben, E.; Sablina, A.; Nuyts, S. p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res. 2014, 74, 1739–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.N.; Cooper, T.S.; Rodriguez, M.; Trummell, H.Q.; Bonner, J.A.; Rosenthal, E.L.; Yang, E.S. DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget 2015, 6, 26995–27007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, Y.; Zheng, Q.; Li, Z.; Fan, F.; Liu, X.; Song, S.; Gu, Y.; Xia, N.; Li, S. [Preparation and Cryo-EM Structure Determination of Human Papillomavirus 16 Pseudovirion Derived from Suspension-adapted HEK293 Cells]. J. Virol. 2016, 32, 551–559. [Google Scholar]
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.K.; Griffith, J.D.; Kreuzer, K.N. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 2007, 67, 8248–8254. [Google Scholar] [CrossRef] [Green Version]
- Friedl, A.A.; Kraxenberger, A.; Eckardt-Schupp, F. An electrophoretic approach to the assessment of the spatial distribution of DNA double-strand breaks in mammalian cells. Electrophoresis 1995, 16, 1865–1874. [Google Scholar] [CrossRef]
- Joshi, N.; Grant, S.G. DNA double-strand break damage and repair assessed by pulsed-field gel electrophoresis. Methods Mol. Biol. 2005, 291, 121–129. [Google Scholar]
- Bryant, H.E. DNA double-strand break damage and repair assessed by pulsed-field gel electrophoresis. Methods Mol. Biol. 2012, 920, 315–321. [Google Scholar] [CrossRef]
- Li, L.H.; Olin, E.J.; Buskirk, H.H.; Reineke, L.M. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 1970, 30, 2760–2769. [Google Scholar]
- Kraakman-van der Zwet, M.; Overkamp, W.J.; van Lange, R.E.; Essers, J.; van Duijn-Goedhart, A.; Wiggers, I.; Swaminathan, S.; van Buul, P.P.; Errami, A.; Tan, R.T.; et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol. Cell. Biol. 2002, 22, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Vispe, S.; DeVries, L.; Creancier, L.; Besse, J.; Breand, S.; Hobson, D.J.; Svejstrup, J.Q.; Annereau, J.P.; Cussac, D.; Dumontet, C.; et al. Triptolide is an inhibitor of RNA polymerase I and II-dependent transcription leading predominantly to down-regulation of short-lived mRNA. Mol. Cancer Ther. 2009, 8, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Koba, M.; Konopa, J. [Actinomycin D and its mechanisms of action]. Postepy Higieny Medycyny Doswiadczalnej 2005, 59, 290–298. [Google Scholar]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xie, R.; Su, J.; Ye, B.; Wei, S.; Liang, Z.; Bai, R.; Chen, Z.; Li, Z.; Gao, X. Inhibition of RNA polymerase III transcription by Triptolide attenuates colorectal tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 217. [Google Scholar] [CrossRef]
- Ross, W.E.; Bradley, M.O. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochimica. Biophysica. Acta 1981, 654, 129–134. [Google Scholar] [CrossRef]
- Andrews, W.J.; Panova, T.; Normand, C.; Gadal, O.; Tikhonova, I.G.; Panov, K.I. Old drug, new target: Ellipticines selectively inhibit RNA polymerase I transcription. J. Biol. Chem. 2013, 288, 4567–4582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourriere, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, P.; Cassel, T.N.; Savolainen, L.; Helleday, T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol. Cell. Biol. 2008, 28, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setton, J.; Lee, N.Y.; Riaz, N.; Huang, S.H.; Waldron, J.; O’Sullivan, B.; Zhang, Z.; Shi, W.; Rosenthal, D.I.; Hutcheson, K.A.; et al. A multi-institution pooled analysis of gastrostomy tube dependence in patients with oropharyngeal cancer treated with definitive intensity-modulated radiotherapy. Cancer 2015, 121, 294–301. [Google Scholar] [CrossRef]
- Dong, Y.; Ridge, J.A.; Ebersole, B.; Li, T.; Lango, M.N.; Churilla, T.M.; Donocoff, K.; Bauman, J.R.; Galloway, T.J. Incidence and outcomes of radiation-induced late cranial neuropathy in 10-year survivors of head and neck cancer. Oral Oncol. 2019, 95, 59–64. [Google Scholar] [CrossRef]
- Xu, B.; Boero, I.J.; Hwang, L.; Le, Q.T.; Moiseenko, V.; Sanghvi, P.R.; Cohen, E.E.; Mell, L.K.; Murphy, J.D. Aspiration pneumonia after concurrent chemoradiotherapy for head and neck cancer. Cancer 2015, 121, 1303–1311. [Google Scholar] [CrossRef]
- Chera, B.S.; Amdur, R.J.; Green, R.; Shen, C.; Gupta, G.; Tan, X.; Knowles, M.; Fried, D.; Hayes, N.; Weiss, J.; et al. Phase II Trial of De-Intensified Chemoradiotherapy for Human Papillomavirus-Associated Oropharyngeal Squamous Cell Carcinoma. J. Clin. Oncol. 2019, 37, 2661–2669. [Google Scholar] [CrossRef]
- Marur, S.; Li, S.; Cmelak, A.J.; Gillison, M.L.; Zhao, W.J.; Ferris, R.L.; Westra, W.H.; Gilbert, J.; Bauman, J.E.; Wagner, L.I.; et al. E1308: Phase II Trial of Induction Chemotherapy Followed by Reduced-Dose Radiation and Weekly Cetuximab in Patients with HPV-Associated Resectable Squamous Cell Carcinoma of the Oropharynx-ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2017, 35, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Hajek, M.; Sewell, A.; Kaech, S.; Burtness, B.; Yarbrough, W.G.; Issaeva, N. TRAF3/CYLD mutations identify a distinct subset of human papilloma virus-associated head and neck squamous cell carcinoma. Cancer 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issaeva, N.; Burtness, B.; Yarbrough, W.G. TRAF3 and CYLD Gene Defects in HPV-Associated Head and Neck Cancer: Biomarker of Response and Indicator of Targets for New Therapies. J. Cancer Clin. Trials 2018, 3, 1000142. [Google Scholar]
- Beaty, B.T.; Moon, D.H.; Shen, C.J.; Amdur, R.J.; Weiss, J.; Grilley-Olson, J.; Patel, S.; Zanation, A.; Hackman, T.G.; Thorp, B.; et al. PIK3CA mutation in HPV-associated OPSCC patients receiving deintensified chemoradiation. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef]
- Gubanova, E.; Brown, B.; Ivanov, S.V.; Helleday, T.; Mills, G.B.; Yarbrough, W.G.; Issaeva, N. Downregulation of SMG-1 in HPV-positive head and neck squamous cell carcinoma due to promoter hypermethylation correlates with improved survival. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Kann, B.H.; Hicks, D.F.; Payabvash, S.; Mahajan, A.; Du, J.; Gupta, V.; Park, H.S.; Yu, J.B.; Yarbrough, W.G.; Burtness, B.A.; et al. Multi-Institutional Validation of Deep Learning for Pretreatment Identification of Extranodal Extension in Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Kann, B.H.; Aneja, S.; Loganadane, G.V.; Kelly, J.R.; Smith, S.M.; Decker, R.H.; Yu, J.B.; Park, H.S.; Yarbrough, W.G.; Malhotra, A.; et al. Pretreatment Identification of Head and Neck Cancer Nodal Metastasis and Extranodal Extension Using Deep Learning Neural Networks. Sci. Rep. 2018, 8, 14036. [Google Scholar] [CrossRef] [Green Version]
- Chera, B.S.; Kumar, S.; Shen, C.; Amdur, R.; Dagan, R.; Green, R.; Goldman, E.; Weiss, J.; Grilley-Olson, J.; Patel, S.; et al. Plasma Circulating Tumor HPV DNA for the Surveillance of Cancer Recurrence in HPV-Associated Oropharyngeal Cancer. J. Clin. Oncol. 2020, 38, 1050–1058. [Google Scholar] [CrossRef]
- Derheimer, F.A.; Hagan, H.M.; Krueger, H.M.; Hanasoge, S.; Paulsen, M.T.; Ljungman, M. RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad. Sci. USA 2007, 104, 12778. [Google Scholar] [CrossRef] [Green Version]
- Daskalakis, M.; Nguyen, T.T.; Nguyen, C.; Guldberg, P.; Kohler, G.; Wijermans, P.; Jones, P.A.; Lubbert, M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 2002, 100, 2957–2964. [Google Scholar] [CrossRef] [Green Version]
- Sugimura, K.; Fukushima, Y.; Ishida, M.; Ito, S.; Nakamura, M.; Mori, Y.; Okumura, K. Cell cycle-dependent accumulation of histone H3.3 and euchromatic histone modifications in pericentromeric heterochromatin in response to a decrease in DNA methylation levels. Exp. Cell Res. 2010, 316, 2731–2746. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowher, H.; Jeltsch, A. Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer Biol. Ther. 2004, 3, 1062–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiianitsa, K.; Maizels, N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res. 2013, 41, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Qian, W.; Zhang, J. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 2012, 13, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Mugal, C.F.; von Grunberg, H.H.; Peifer, M. Transcription-induced mutational strand bias and its effect on substitution rates in human genes. Mol. Biol. Evol. 2009, 26, 131–142. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, G.; d’Adda di Fagagna, F. Transcription and DNA Damage: Holding Hands or Crossing Swords? J. Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Duch, A.; de Nadal, E.; Posas, F. Dealing with transcriptional outbursts during S phase to protect genomic integrity. J. Mol. Biol. 2013, 425, 4745–4755. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A.; Gaillard, H. Transcription and recombination: When RNA meets DNA. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing replication stress to maintain genome stability: Resolving conflicts between replication and transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA: RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajek, M.; Biktasova, A.; Sewell, A.; Gary, C.; Cantalupo, P.; Anderson, K.S.; Yarbrough, W.G.; Issaeva, N. Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells. Cancers 2021, 13, 21. https://doi.org/10.3390/cancers13010021
Hajek M, Biktasova A, Sewell A, Gary C, Cantalupo P, Anderson KS, Yarbrough WG, Issaeva N. Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells. Cancers. 2021; 13(1):21. https://doi.org/10.3390/cancers13010021
Chicago/Turabian StyleHajek, Michael, Asel Biktasova, Andrew Sewell, Cyril Gary, Paul Cantalupo, Karen S. Anderson, Wendell G. Yarbrough, and Natalia Issaeva. 2021. "Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells" Cancers 13, no. 1: 21. https://doi.org/10.3390/cancers13010021
APA StyleHajek, M., Biktasova, A., Sewell, A., Gary, C., Cantalupo, P., Anderson, K. S., Yarbrough, W. G., & Issaeva, N. (2021). Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV-Associated Head and Neck Cancer Cells. Cancers, 13(1), 21. https://doi.org/10.3390/cancers13010021