The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities


Abstract
:Simple Summary
Abstract
1. Introduction
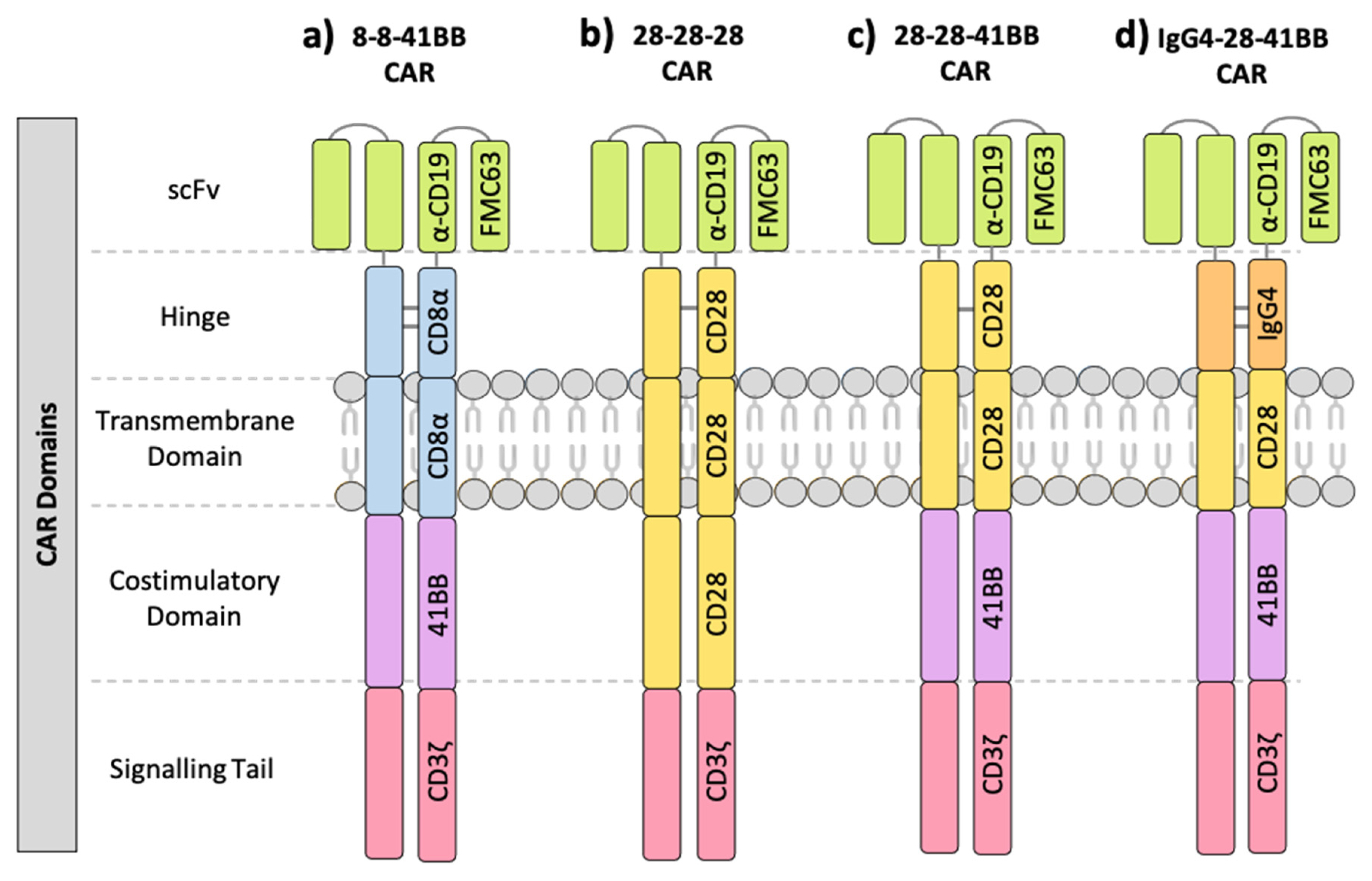
1.1. Components of the Chimeric Antigen Receptor (CAR)
1.2. Limitations of CAR-T Cell Therapy
2. Clinical Trials of Anti-CD19 CAR-T Cell Therapy
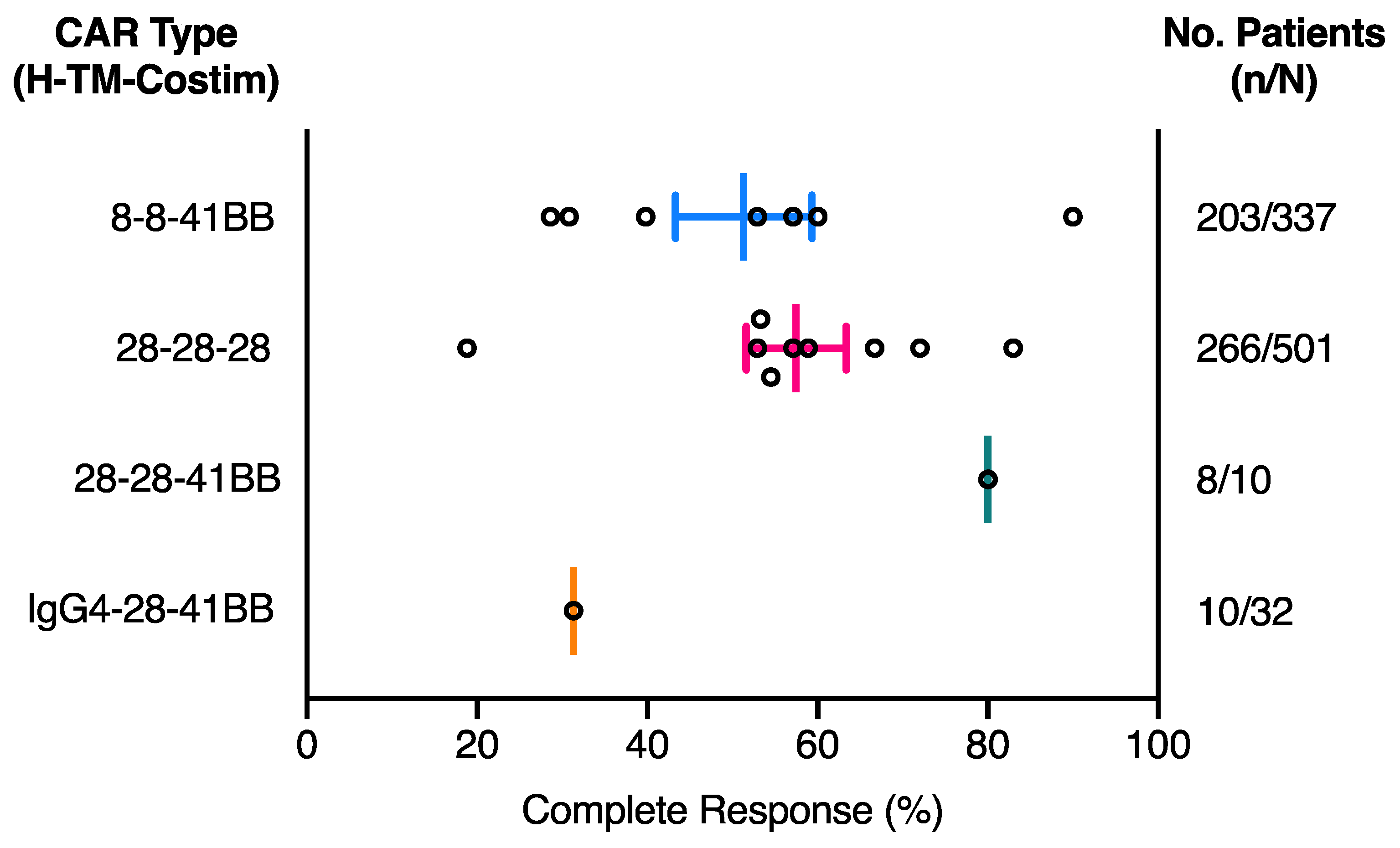
2.1. The Effect of CAR Domains on Clinical Response
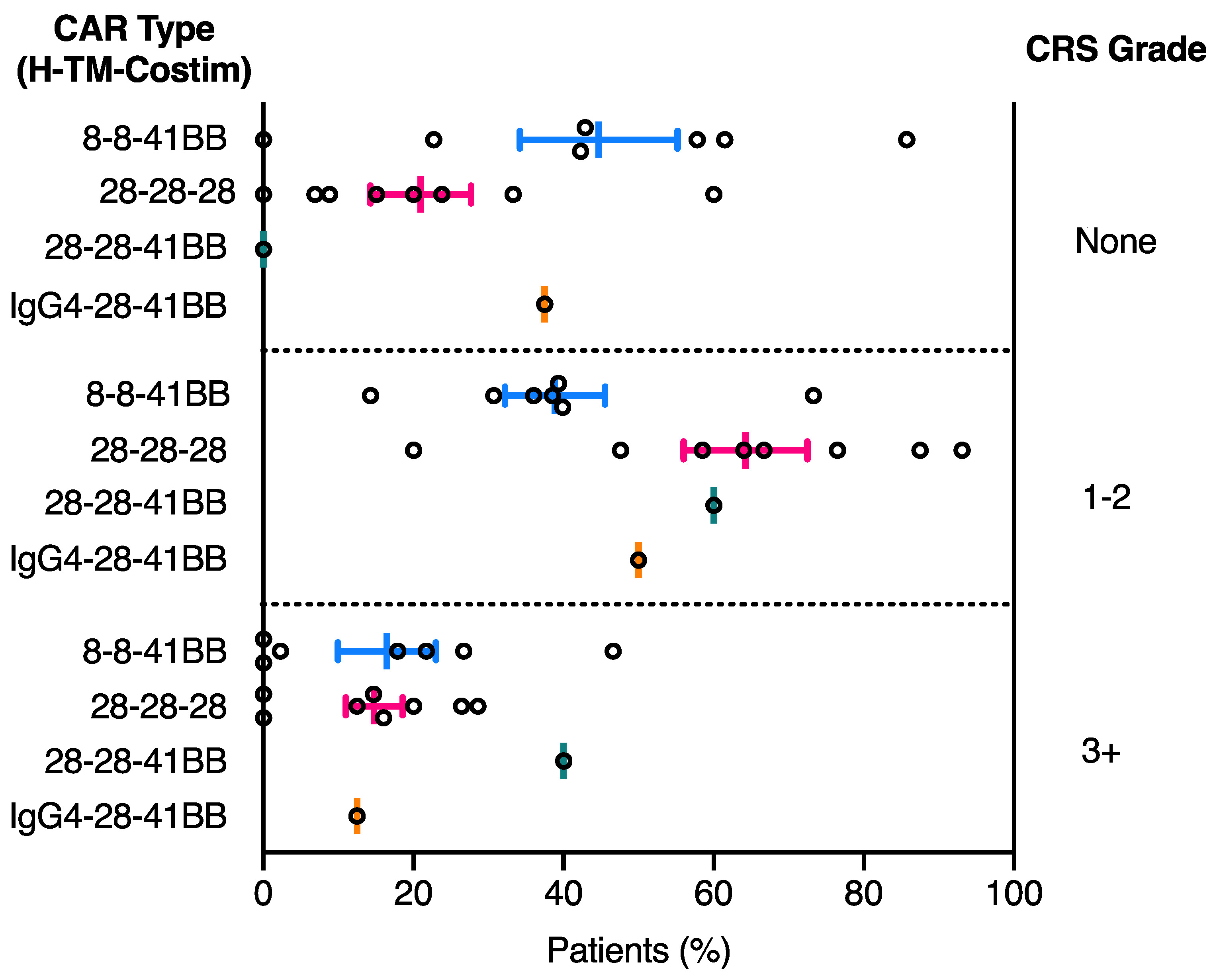
2.2. CRS Toxicity Correlates with CAR Domain Design
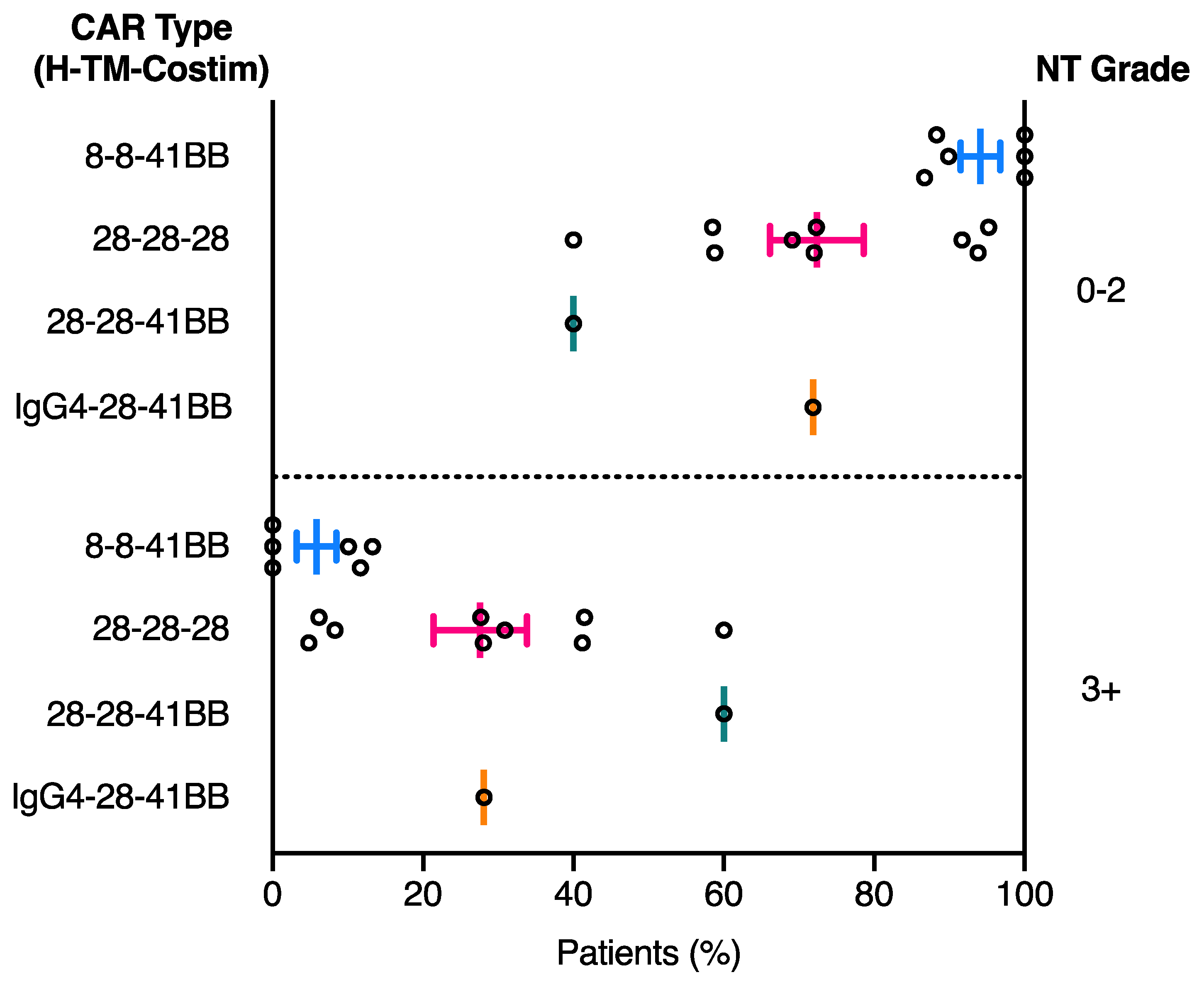
2.3. Relationship between CAR Domain Structure and Neurotoxicity Development
3. Preclinical Evidence of CAR Domain Design Influencing CAR-Related Toxicities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transpl. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Moritz, D.; Groner, B. A spacer region between the single chain antibody- and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther. 1995, 2, 539–546. [Google Scholar]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Leddon, S.A.; Fettis, M.M.; Abramo, K.; Kelly, R.; Oleksyn, D.; Miller, J. The CD28 Transmembrane Domain Contains an Essential Dimerization Motif. Front. Immunol. 2020, 11, 1519. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the antigen density requirement for CAR T cell activity. Cancer Discov. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.J.; Vinanica, N.; Campana, D. Chimeric antigen receptor-T cells with cytokine neutralizing capacity. Blood Adv. 2020, 4, 1419–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine release syndrome after chimeric antigen receptor T Cell therapy for acute lymphoblastic leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Prudent, V.; Breitbart, W.S. Chimeric antigen receptor T-cell neuropsychiatric toxicity in acute lymphoblastic leukemia. Palliat. Support. Care 2017, 15, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Belin, C.; Devic, P.; Ayrignac, X.; Dos Santos, A.; Paix, A.; Sirven-Villaros, L.; Simard, C.; Lamure, S.; Gastinne, T.; Ursu, R.; et al. Description of neurotoxicity in a series of patients treated with CAR T-cell therapy. Sci. Rep. 2020, 10, 18997. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell 2020, 183, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Nadler, L.M.; Anderson, K.C.; Marti, G.; Bates, M.; Park, E.; Daley, J.F.; Schlossman, S.F. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J. Immunol. 1983, 131, 244–250. [Google Scholar] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, I.C.; Lenton, K.A.; Little, D.J.; Decorso, T.; Lee, F.T.; Scott, A.M.; Zola, H.; Hohmann, A.W. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 1997, 34, 1157–1165. [Google Scholar] [CrossRef]
- Ma, F.; Ho, J.Y.; Du, H.; Xuan, F.; Wu, X.; Wang, Q.; Wang, L.; Liu, Y.; Ba, M.; Wang, Y.; et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor-modified T cells in a phase i study targeting pediatrics with acute lymphoblastic leukemia. Hematol. Oncol. 2019, 37, 601–608. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B bell lymphomas. Blood 2019, 134, 241. [Google Scholar] [CrossRef]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Hollyman, D.; Stefanski, J.; Przybylowski, M.; Bartido, S.; Borquez-Ojeda, O.; Taylor, C.; Yeh, R.; Capacio, V.; Olszewska, M.; Hosey, J.; et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J. Immunother. 2009, 32, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, C.S.; Senechal, B.; Riviere, I.; Ni, A.; Bernal, Y.; Wang, X.; Purdon, T.; Hall, M.; Singh, A.N.; Szenes, V.Z.; et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2019, 134, 626–635. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971. [Google Scholar] [CrossRef] [Green Version]
- Drokow, E.K.; Ahmed, H.A.W.; Amponsem-Boateng, C.; Akpabla, G.S.; Song, J.; Shi, M.; Sun, K. Survival outcomes and efficacy of autologous CD19 chimeric antigen receptor-T cell therapy in the patient with diagnosed hematological malignancies: A systematic review and meta-analysis. Clin. Risk Manag. 2019, 15, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Wing, M.G.; Moreau, T.; Greenwood, J.; Smith, R.M.; Hale, G.; Isaacs, J.; Waldmann, H.; Lachmann, P.J.; Compston, A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Investig. 1996, 98, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef] [PubMed]
- Services, U.D.o.H.a.H. Common Terminology Criteria for Adverse Events (CTCAE). V4.03. Available online: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf (accessed on 12 November 2020).
- Services, U.D.o.H.a.H. Common Terminology Criteria for Adverse Events (CTCAE). V3.0. Available online: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf (accessed on 12 November 2020).
- Winkler, U.; Jensen, M.; Manzke, O.; Schulz, H.; Diehl, V.; Engert, A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999, 94, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transpl. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maziarz, R.T.; Schuster, S.J.; Romanov, V.V.; Rusch, E.S.; Li, J.; Signorovitch, J.E.; Maloney, D.G.; Locke, F.L. Grading of neurological toxicity in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020, 4, 1440–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Lam, N.; Vanasse, D.; Shen, Y.W.; Rose, J.J.; Rossi, J.; Xue, A.; Bot, A.; Scholler, N.; Mikkilineni, L.; et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020, 26, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.D.; Nguyen, D.P.; Ferreira, L.M.R.; Ho, P.; Raffin, C.; Valencia, R.B.; Congrave-Wilson, Z.; Roth, T.; Eyquem, J.; Van Gool, F.; et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. bioRxiv 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT ID | Tumour Type | No. of Patients | CAR Name | CAR Domain Structure | Gene Transfer Method | CAR-T Cell Dose | Ref | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ScFv | Hinge | TMD | Costim | Signal | |||||||
| NCT02842138 | FL | 7 | CD19-BBz(86) | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 3 × 108–3.7 × 108 | [15] |
| NCT02842138 | DLBCL | 13 | CD19-BBz(86) | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 6 × 106–3.2 × 108 | [15] |
| NCT02030834 | DLBCL, FL | 28 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 3.1 × 106–8.9 × 106/kg | [39] |
| NCT02445248 | DLBCL | 111 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 1 × 107–6 × 108 | [40] |
| NCT01626495 NCT01029366 | B-ALL, T-ALL | 30 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 1 × 107–1 × 108/kg | [30,41] |
| NCT02435849 | B-ALL | 75 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 3.1 × 106/kg | [42] |
| NCT02631044 | DLBCL | 268 | JCAR017 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | lentivirus | 5 × 107–1.5 × 108 | [43] |
| NCT01860937 | B-ALL | 25 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 1 × 106–3 × 106/kg | [44] |
| NCT01044069 | B-ALL | 53 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 1 × 106–3 × 106/kg | [23,45] |
| NCT00466531 | CLL | 16 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 2.6 × 106–3.2 × 107 | [46] |
| NCT01840566 | DLBCL, FL, MZL, MCL | 15 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 5 × 106–1 × 107/kg | [47] |
| NCT02601313 | MCL | 68 | KTE-X19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 2 × 106/kg | [48] |
| NCT02348216 | DLBCL, PMBCL, TFL | 101 | KTE-C19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 2 × 106/kg | [49] |
| NCT00924326 | DLBCL | 17 | CAR-19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 1 × 106–2 × 106/kg | [50] |
| NCT01593696 | B-ALL | 21 | FMC63-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | gamma retrovirus | 3 × 104–3 × 106/kg | [51] |
| NCT02963038 | B-ALL | 10 | SENL-B19 | FMC63 | CD28 | CD28 | 41BB | CD3ζ | lentivirus | 3 × 105–1.6 × 106/kg | [36] |
| NCT01865617 | LBCL, FL, MCL | 32 | - | FMC63 | IgG4 | CD28 | 41BB | CD3ζ | lentivirus | 2 × 105–2 × 107/kg | [37,38] |
| Prognostic Factor | No. of Patients | No. of Clinical Trials | Response Rate | p-Value |
|---|---|---|---|---|
| Mean % [95% CI] | ||||
| Age: | 0.7743 | |||
| 65 | 483 | 12 | 50.7% [39.1–62.3] | |
| >65 | 180 | 5 | 52.6% [24.0–81.1] | |
| Lymphodepletion: | 0.4737 | |||
| Yes | 835 | 12 | 57.1% [46.9–67.4] | |
| No | 13 | 3 | 42.9% [0–100] | |
| CAR-T cells infused: | 0.2043 | |||
| 1 × 107/kg | 442 | 12 | 58.5% [47.0–70.0] | |
| >1 × 107/kg | 35 | 5 | 43.4% [5.8–81.0] | |
| Cancer type: | ||||
| B-ALL | 184 | 5 | 72.3% [60.6–84.1] | |
| DLBCL | 490 | 9 | 41.4% [32.6–50.2] | |
| FL | 49 | 5 | 52.5% [30.2–74.7] | |
| MCL | 73 | 3 | 22.2% [0–100] |
| NCT ID | CAR Name | CAR Domain Structure | No. of Patients | CRS Grading System | Neurotoxicity Grading System | ||||
|---|---|---|---|---|---|---|---|---|---|
| ScFv | Hinge | TM | Costim | Signal | |||||
| NCT02842138 | CD19-BBz(86) | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 7 | CTCAE v4.03 [57] | CTCAE v4.03 [57] |
| NCT02842138 | CD19-BBz(86) | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 13 | CTCAE v4.03 [57] | CTCAE v4.03 [57] |
| NCT02030834 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 28 | Penn grading system [41] | CTCAE v3.0 [58] |
| NCT02445248 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 111 | Penn grading system [41] | CTCAE v4.03 [57] |
| NCT01626495 NCT01029366 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 30 | Penn grading system [41] | CTCAE v3.0 [58] |
| NCT02435849 | CTL-019 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 75 | MedDRA and CTCAE v4.03 [57] | MedDRA and CTCAE v4.03 [57] |
| NCT02631044 | JCAR017 | FMC63 | CD8α | CD8α | 41BB | CD3ζ | 268 | Lee et al. 2014 [60] | CTCAE v4.03 [57] |
| NCT01860937 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 25 | CTCAE v4.0 | CTCAE v4.0 |
| NCT01044069 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 53 | Lee et al. 2014 [60] | CTCAE v4.03 [57] |
| NCT00466531 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 16 | CTCAE v3.0 (<2009) [58] CTCAE v4.0 (>2009) | CTCAE v3.0 (<2009) [58] CTCAE v4.0 (>2009) |
| NCT01840566 | 19-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 15 | ASBMT | CTCAE v4.03 [57] |
| NCT02601313 | KTE-X19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 68 | Lee et al. 2014 [60] | CTCAE v4.03 [57] |
| NCT02348216 | KTE-C19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 101 | Lee et al. 2014 [60] | CTCAE v4.03 [57] |
| NCT00924326 | CAR-19 | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 17 | CTCAE v3.0 [58] | CTCAE v3.0 [58] |
| NCT01593696 | FMC63-28z | FMC63 | CD28 | CD28 | CD28 | CD3ζ | 21 | CTCAE v4.02 | CTCAE v4.02 |
| NCT02963038 | SENL-B19 | FMC63 | CD28 | CD28 | 41BB | CD3ζ | 10 | CTCAE v4.0 | CTCAE v4.0 |
| NCT01865617 | - | FMC63 | IgG4 | CD28 | 41BB | CD3ζ | 32 | CTCAE v4.03 [57] | CTCAE v4.03 [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davey, A.S.; Call, M.E.; Call, M.J. The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities. Cancers 2021, 13, 38. https://doi.org/10.3390/cancers13010038
Davey AS, Call ME, Call MJ. The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities. Cancers. 2021; 13(1):38. https://doi.org/10.3390/cancers13010038
Chicago/Turabian StyleDavey, Ashleigh S., Matthew E. Call, and Melissa J. Call. 2021. "The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities" Cancers 13, no. 1: 38. https://doi.org/10.3390/cancers13010038
APA StyleDavey, A. S., Call, M. E., & Call, M. J. (2021). The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities. Cancers, 13(1), 38. https://doi.org/10.3390/cancers13010038