
CD137 as an Attractive T Cell Co-Stimulatory Target in the TNFRSF for Immuno-Oncology Drug Development


Abstract
:Simple Summary
Abstract
1. Introduction
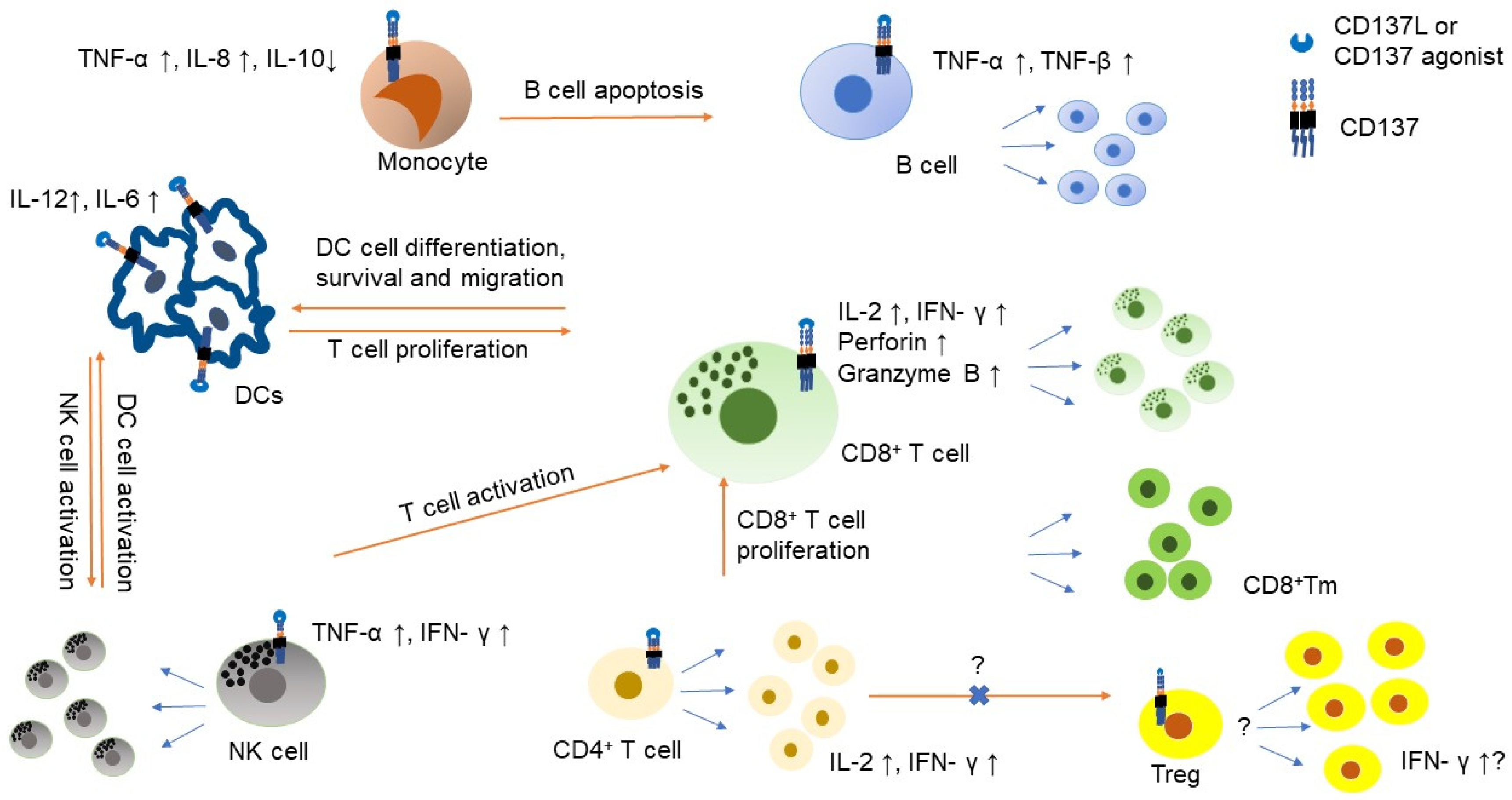
2. The Role of CD137 in the Tumor Microenvironment
2.1. Prevalence of CD137-Positive T Cells in Tumor Microenvironment
2.2. The Role of CD137 in the Context of Clincial Trials
2.2.1. CD27
2.2.2. OX40
2.2.3. GITR
2.2.4. CD137
2.3. The Role of CD137 in Other Immune Cells
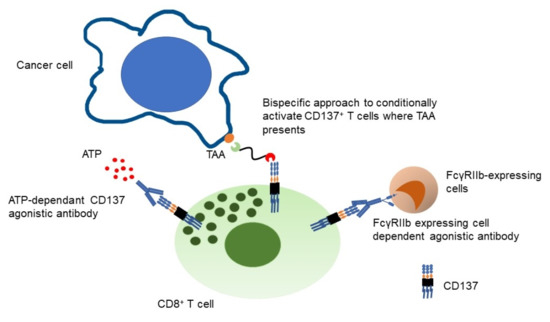
3. Technological Advances to Mitigate Hepatotoxicity for CD137 Targeting Molecules
3.1. Bispecific Molecules Targeting CD137
3.2. Agonistic Monoclonal Antibodies to CD137 with an Extra Safety Switch
4. Potential Combination Partners for CD137 Targeting Molecules
4.1. Checkpoint Inhibitors
4.2. CD3 Targeting Bispecific Compounds
4.3. Anti-Angiogenic Compounds
4.4. ADCC Enhanced Antibodies
5. Discussion
Challenges of CD137 Targeting Molecules in Solid Tumors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwon, B.S.; Weissman, S.M. cDNA sequences of two inducible T-cell genes. Proc. Natl. Acad. Sci. USA 1989, 86, 1963–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollok, K.E.; Kim, Y.J.; Zhou, Z.; Hurtado, J.; Kim, K.K.; Pickard, R.T.; Kwon, B.S. Inducible T cell antigen 4-1BB. Analysis of expression and function. J. Immunol. 1993, 150, 771–781. [Google Scholar]
- Garni-Wagner, B.A.; Lee, Z.H.; Kim, Y.J.; Wilde, C.; Kang, C.Y.; Kwon, B.S. 4-1BB is expressed on CD45RAhiROhi transitional T cell in humans. Cell. Immunol. 1996, 169, 91–98. [Google Scholar] [CrossRef]
- Schwarz, H.; Valbracht, J.; Tuckwell, J.; von Kempis, J.; Lotz, M. ILA, the human 4-1BB homologue, is inducible in lymphoid and other cell lineages. Blood 1995, 85, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Kwon, B.S. 4-1BB signaling beyond T cells. Cell. Mol. Immunol. 2011, 8, 281–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, H.; Arden, K.; Lotz, M. CD137, a member of the tumor necrosis factor receptor family, is located on chromosome 1p36, in a cluster of related genes, and colocalizes with several malignancies. Biochem. Biophys. Res. Commun. 1997, 235, 699–703. [Google Scholar] [CrossRef]
- Ward-Kavanagh, L.K.; Lin, W.W.; Sedy, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [Green Version]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Chin, S.M.; Kimberlin, C.R.; Roe-Zurz, Z.; Zhang, P.; Xu, A.; Liao-Chan, S.; Sen, D.; Nager, A.R.; Oakdale, N.S.; Brown, C.; et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat. Commun. 2018, 9, 4679. [Google Scholar] [CrossRef] [PubMed]
- Wortzman, M.E.; Clouthier, D.L.; McPherson, A.J.; Lin, G.H.; Watts, T.H. The contextual role of TNFR family members in CD8(+) T-cell control of viral infections. Immunol. Rev. 2013, 255, 125–148. [Google Scholar] [CrossRef]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Schwartz, R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987, 165, 302–319. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Wilcox, R.A.; Tamada, K.; Strome, S.E.; Chen, L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J. Immunol. 2002, 169, 4230–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, J.E.; Kerr, J.P.; Rogel, A.; Taraban, V.Y.; Buchan, S.L.; Johnson, P.W.; Al-Shamkhani, A. Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J. Immunol. 2014, 193, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Akhmetzyanova, I.; Zelinskyy, G.; Littwitz-Salomon, E.; Malyshkina, A.; Dietze, K.K.; Streeck, H.; Brandau, S.; Dittmer, U. CD137 Agonist Therapy Can Reprogram Regulatory T Cells into Cytotoxic CD4+ T Cells with Antitumor Activity. J. Immunol. 2016, 196, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Gao, F.; Wang, Q.; Wang, X.; Zhu, F.; Ma, C.; Sun, W.; Zhang, L. Agonistic anti-4-1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand. J. Immunol. 2007, 66, 435–440. [Google Scholar] [CrossRef]
- Madireddi, S.; Schabowsky, R.H.; Srivastava, A.K.; Sharma, R.K.; Yolcu, E.S.; Shirwan, H. SA-4-1BBL costimulation inhibits conversion of conventional CD4+ T cells into CD4+ FoxP3+ T regulatory cells by production of IFN-gamma. PLoS ONE 2012, 7, e42459. [Google Scholar] [CrossRef]
- Futagawa, T.; Akiba, H.; Kodama, T.; Takeda, K.; Hosoda, Y.; Yagita, H.; Okumura, K. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002, 14, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Voskens, C.J.; Sallin, M.; Maniar, A.; Montes, C.L.; Zhang, Y.; Lin, W.; Li, G.; Burch, E.; Tan, M.; et al. CD137 promotes proliferation and survival of human B cells. J. Immunol. 2010, 184, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J., Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.D.; Park, S.; Jeong, S.; Lee, Y.J.; Lee, H.; Kim, C.G.; Kim, K.H.; Hong, S.M.; Lee, J.Y.; Kim, S.; et al. 4-1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8(+) T Cells in Hepatocellular Carcinoma. Hepatology 2020, 71, 955–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M. The TNF family in T cell differentiation and function--unanswered questions and future directions. Semin. Immunol. 2014, 26, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulle, G.; Vidric, M.; Watts, T.H. IL-15-dependent induction of 4-1BB promotes antigen-independent CD8 memory T cell survival. J. Immunol. 2006, 176, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.F.; Soroosh, P.; Takahashi, T.; Yoshikai, Y.; Shen, H.; Lefrancois, L.; Borst, J.; Sugamura, K.; Ishii, N. OX40 costimulatory signals potentiate the memory commitment of effector CD8+ T cells. J. Immunol. 2008, 181, 5990–6001. [Google Scholar] [CrossRef] [Green Version]
- Gaspal, F.M.; Kim, M.Y.; McConnell, F.M.; Raykundalia, C.; Bekiaris, V.; Lane, P.J. Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. J. Immunol. 2005, 174, 3891–3896. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, J.; Xiao, Y.; Rossen, J.W.; van der Sluijs, K.F.; Sugamura, K.; Ishii, N.; Borst, J. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J. Immunol. 2005, 175, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Salek-Ardakani, S.; Song, J.; Halteman, B.S.; Jember, A.G.; Akiba, H.; Yagita, H.; Croft, M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J. Exp. Med. 2003, 198, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Soroosh, P.; Doherty, T.A.; So, T.; Mehta, A.K.; Khorram, N.; Norris, P.S.; Scheu, S.; Pfeffer, K.; Ware, C.; Croft, M. Herpesvirus entry mediator (TNFRSF14) regulates the persistence of T helper memory cell populations. J. Exp. Med. 2011, 208, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Flinn, I.; Taylor, M.H.; Sikic, B.I.; Brody, J.; Nemunaitis, J.; Feldman, A.; Hawthorne, T.R.; Rawls, T.; Keler, T.; et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic malignancies. Blood Adv. 2020, 4, 1917–1926. [Google Scholar] [CrossRef]
- Shapira-Frommer, R.; van Dongen, M.G.; Dobrenkov, K.; Chartash, E.; Liu, F.; Li, C.; Wnek, R.; Patel, M. O83 Phase 1 study of an anti-CD27 agonist as monotherapy and in combination with pembrolizumab in patients with advanced solid tumors. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Spano, J.-P.; Angevin, E.; Doi, T.; Bullock, A.J.; Harris, W.P.; Hamid, O.; Gougis, P.; Forgie, A.; Yang, W.; et al. Analysis of OX40 agonist antibody (PF-04518600) in patients with hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 523. [Google Scholar] [CrossRef]
- Glisson, B.S.; Leidner, R.S.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; Schneider, R.; Goel, S.; Ohr, J.P.; Burton, J.; et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 5358–5367. [Google Scholar] [CrossRef]
- Duhen, R.; Ballesteros-Merino, C.; Frye, A.K.; Tran, E.; Rajamanickam, V.; Chang, S.C.; Koguchi, Y.; Bifulco, C.B.; Bernard, B.; Leidner, R.S.; et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat. Commun. 2021, 12, 1047. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Lam, V.K.; Ros, W.; Bauer, T.M.; Hansen, A.R.; Cho, D.C.; Hodi, F.S.; Schellens, J.H.M.; Litton, J.K.; Aspeslagh, S.; et al. Abstract CT150: A first-in-human phase I study of the OX40 agonist GSK3174998 (GSK998) +/− pembrolizumab in patients (Pts) with selected advanced solid tumors (ENGAGE-1). Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Infante, J.R.; Hansen, A.R.; Pishvaian, M.J.; Chow, L.Q.M.; McArthur, G.A.; Bauer, T.M.; Liu, S.V.; Sandhu, S.K.; Tsai, F.Y.-C.; Kim, J.; et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 101. [Google Scholar] [CrossRef]
- Moiseyenko, A.; Muggia, F.; Condamine, T.; Pulini, J.; Janik, J.E.; Cho, D.C. Sequential therapy with INCAGN01949 followed by ipilimumab and nivolumab in two patients with advanced ovarian carcinoma. Gynecol. Oncol. Rep. 2020, 34, 100655. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Moreno, V.; Heinhuis, K.M.; Olszanski, A.J.; Spreafico, A.; Ong, M.; Chu, Q.; Carvajal, R.D.; Trigo, J.; Ochoa de Olza, M.; et al. OX40 Agonist BMS-986178 Alone or in Combination With Nivolumab and/or Ipilimumab in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Autio, K.; Golan, T.; Dobrenkov, K.; Chartash, E.; Chen, Q.; Wnek, R.; Long, G.V. Phase I Study of MK-4166, an Anti-human Glucocorticoid-Induced TNF Receptor Antibody, Alone or with Pembrolizumab in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Geva, R.; Voskoboynik, M.; Dobrenkov, K.; Mayawala, K.; Gwo, J.; Wnek, R.; Chartash, E.; Long, G.V. First-in-human phase 1 study of MK-1248, an anti-glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors. Cancer 2020, 126, 4926–4935. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; Di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination With Nivolumab for Patients With Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2019, 6, 100–107. [Google Scholar]
- Balmanoukian, A.S.; Infante, J.R.; Aljumaily, R.; Naing, A.; Chintakuntlawar, A.V.; Rizvi, N.A.; Ross, H.J.; Gordon, M.; Mallinder, P.R.; Elgeioushi, N.; et al. Safety and Clinical Activity of MEDI1873, a Novel GITR Agonist, in Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 6196–6203. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Carvajal, R.D.; Marabelle, A.; Patel, S.P.; LoRusso, P.M.; Rasmussen, E.; Juan, G.; Upreti, V.V.; Beers, C.; Ngarmchamnanrith, G.; et al. Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor-related protein agonist AMG 228 in patients with advanced solid tumors. J. Immunother Cancer 2018, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, J.; Herbaux, C.; Ribrag, V.; Zelenetz, A.D.; Houot, R.; Neelapu, S.S.; Logan, T.; Lossos, I.S.; Urba, W.; Salles, G.; et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am. J. Hematol. 2020, 95, 510–520. [Google Scholar] [CrossRef]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2020, 4, e000629. [Google Scholar] [CrossRef] [Green Version]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [Green Version]
- Diab, A. A first-in-human (FIH) study of PF-04518600 (PF-8600) OX40 agonist in adult patients (pts) with select advanced malignancies. Ann. Oncol. 2016, 27, 359–378. [Google Scholar] [CrossRef]
- Goldman, J.W.; Piha-Paul, S.A.; Curti, B.D.; Pedersen, K.; Bauer, T.M.; Groenland, S.L.; Carvajal, R.D.; Chhaya, V.; Hammond, S.A.; Streicher, K.; et al. Safety and tolerability of MEDI0562 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3003. [Google Scholar] [CrossRef]
- Oberst, M.D.; Auge, C.; Morris, C.; Kentner, S.; Mulgrew, K.; McGlinchey, K.; Hair, J.; Hanabuchi, S.; Du, Q.; Damschroder, M.; et al. Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Mol. Cancer Ther. 2018, 17, 1024–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappori, A.; Thompson, J.; Eskens, F.; Spano, J.-P.; Doi, T.; Hamid, O.; Diab, A.; Rizvi, N.; Hu-Lieskovan, S.; Ros, W.; et al. Results from a combination of OX40 (PF-04518600) and 4–1BB (utomilumab) agonistic antibodies in melanoma and non-small cell lung cancer in a phase 1 dose expansion cohort. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Sznol, M.; Hodi, F.S.; Margolin, K.; McDermott, D.F.; Ernstoff, M.S.; Wojtaszek, J.M.K.; Feltquate, D.; Logan, T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J. Clin. Oncol. 2008, 26, 3007. [Google Scholar] [CrossRef]
- Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Chin, R.; Chen, C.H.; Budhani, P.; Ai, M.; Reilley, M.J.; Sebastian, M.M.; Hong, D.S.; et al. Activation of 4-1BB on Liver Myeloid Cells Triggers Hepatitis via an Interleukin-27-Dependent Pathway. Clin. Cancer Res. 2018, 24, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Judkins, C.; Hoare, J.; Klein, R.; Parkinson, R.; Wang, H.; Cao, H.; Durham, J.; Purtell, K.; De Jesus-Acosta, A.; et al. Urelumab (anti-CD137 agonist) in combination with vaccine and nivolumab treatments is safe and associated with pathologic response as neoadjuvant and adjuvant therapy for resectable pancreatic cancer. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Hurtado, J.C.; Kim, S.H.; Pollok, K.E.; Lee, Z.H.; Kwon, B.S. Potential role of 4-1BB in T cell activation. Comparison with the costimulatory molecule CD28. J. Immunol. 1995, 155, 3360–3367. [Google Scholar]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef] [Green Version]
- You, G.; Lee, Y.; Kang, Y.W.; Park, H.W.; Park, K.; Kim, H.; Kim, Y.M.; Kim, S.; Kim, J.H.; Moon, D.; et al. B7-H3x4-1BB bispecific antibody augments antitumor immunity by enhancing terminally differentiated CD8(+) tumor-infiltrating lymphocytes. Sci. Adv. 2021, 7, eaax3160. [Google Scholar] [CrossRef]
- Wilcox, R.A.; Chapoval, A.I.; Gorski, K.S.; Otsuji, M.; Shin, T.; Flies, D.B.; Tamada, K.; Mittler, R.S.; Tsuchiya, H.; Pardoll, D.M.; et al. Cutting edge: Expression of functional CD137 receptor by dendritic cells. J. Immunol. 2002, 168, 4262–4267. [Google Scholar] [CrossRef]
- Dharmadhikari, B.; Wu, M.; Abdullah, N.S.; Rajendran, S.; Ishak, N.D.; Nickles, E.; Harfuddin, Z.; Schwarz, H. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology 2016, 5, e1113367. [Google Scholar] [CrossRef]
- Kwajah, M.M.S.; Schwarz, H. CD137 ligand signaling induces human monocyte to dendritic cell differentiation. Eur J. Immunol. 2010, 40, 1938–1949. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.S.; Tang, T.; Jia, J.L.; Xie, B.C.; Wu, T.L.; Sheng, Y.Y.; Xue, Y.Z.; Tang, H.M. CD137 agonist induces gastric cancer cell apoptosis by enhancing the functions of CD8(+) T cells via NF-kappaB signaling. Cancer Cell Int. 2020, 20, 513. [Google Scholar] [CrossRef]
- Bacher, P.; Heinrich, F.; Stervbo, U.; Nienen, M.; Vahldieck, M.; Iwert, C.; Vogt, K.; Kollet, J.; Babel, N.; Sawitzki, B.; et al. Regulatory T Cell Specificity Directs Tolerance versus Allergy against Aeroantigens in Humans. Cell 2016, 167, 1067–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachapati, K.; Adams, D.E.; Wu, Y.; Steward, C.A.; Rainbow, D.B.; Wicker, L.S.; Mittler, R.S.; Ridgway, W.M. The B10 Idd9.3 locus mediates accumulation of functionally superior CD137(+) regulatory T cells in the nonobese diabetic type 1 diabetes model. J. Immunol. 2012, 189, 5001–5015. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhu, G.; Luo, L.; Flies, A.S.; Chen, L. CD137 stimulation delivers an antigen-independent growth signal for T lymphocytes with memory phenotype. Blood 2007, 109, 4882–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.; Miller, R.; Nilsson, A.; Ljung, L.; Chunyk, A.; McMahan, C.; Bienvenue, D.; Askmyr, M.; Gabriela Hernandez-Hoyos, G.; Fritzell, S. Potent tumor-directed T cell activation and in vivo tumor inhibition induced by a 4–1BB x 5T4 ADAPTIR™ bispecific antibody. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.B.; Horton, B.L.; Zheng, Y.; Duan, Y.; Powell, J.D.; Gajewski, T.F. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med. 2017, 214, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; Bolanos, E.; Teijeira, A.; Martinez-Forero, I.; Labiano, S.; Azpilikueta, A.; Morales-Kastresana, A.; Quetglas, J.I.; Wagena, E.; Sanchez-Paulete, A.R.; et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Ku, G.; Bendell, J.C.; Tolcher, A.W.; Hurvitz, S.A.; Krishnamurthy, A.; El-Khoueiry, A.B.; Patnaik, A.; Shroff, R.T.; Noonan, A.; Hahn, N.M.; et al. A phase I dose escalation study of PRS-343, a HER2/4-1BB bispecific molecule, in patients with HER2-positive malignancies. Ann. Oncol. 2020, 31, 462–504. [Google Scholar] [CrossRef]
- Piha-Paul, S. Phase 1 Dose Escalation Study of PRS-343, a HER2/4-1BB Bispecific Molecule, in Patients with HER2+ Malignancies. In Proceedings of the 34th Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer, National Harbor, MD, USA, 6–10 November 2019. [Google Scholar]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- Garralda, E.; Geva, R.; Ben-Ami, E.; Maurice-Dror, C.; Calvo, E.; LoRusso, P.; Türeci, Ö.; Niewood, M.; Şahin, U.; Jure-Kunkel, M.; et al. 412 First-in-human phase I/IIa trial to evaluate the safety and initial clinical activity of DuoBody®-PD-L1×4–1BB (GEN1046) in patients with advanced solid tumors. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Husser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11, eaav5989. [Google Scholar] [CrossRef]
- Melero, I.; Sanmamed, M.F.; Calvo, E.; Moreno, V.; Hernandez Guerrero, T.C.; Martinez-Garcia, M.; Rodriguez-Vid, A.; Tabernero, J.; Azaro Pedrazzoli, A.B.; Spanggaard, I.; et al. First-in-human (FIH) phase I study of RO7122290 (RO), a novel FAP-targeted 4-1BB agonist, administered as single agent and in combination with atezolizumab (ATZ) to patients with advanced solid. Ann. Oncol. 2020, 31, S707. [Google Scholar] [CrossRef]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 Activated by Extracellular Adenosine Triphosphate Is Tumor Selective and Broadly Effective In Vivo without Systemic Immune Activation. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Galand, C.; Venkatraman, V.; Marques, M.; Strauss, J.; Carvajal, R.; Lim, M.; Morin, B.; Ignatovich, O.; Findeis, M.; Underwood, D.; et al. 377 AGEN2373 is a CD137 agonist antibody designed to leverage optimal CD137 and FcγR co-targeting to promote antitumor immunologic effects. J. ImmunoTher. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Qi, X.; Li, F.; Wu, Y.; Cheng, C.; Han, P.; Wang, J.; Yang, X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nat. Commun. 2019, 10, 2141. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Repasky, E.A.; Wood, L.S.; Butterfield, L.H. Highlights of the 31st annual meeting of the Society for Immunotherapy of Cancer (SITC), 2016. J. Immunother. Cancer 2017, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-cell and NK-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Levy, R.; Houot, R.; Patel, S.P.; Popplewell, L.; Jacobson, C.; Mu, X.J.; Deng, S.; Ching, K.A.; Chen, Y.; et al. First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20(+) Non-Hodgkin Lymphomas. Clin. Cancer Res. 2020, 26, 2524–2534. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Rahimizadeh, P.; Yang, S.; Lim, S.I. Albumin: An Emerging Opportunity in Drug Delivery. Biotechnol. Bioprocess. Eng. Vol. 2020, 25, 985–995. [Google Scholar] [CrossRef]
- Nessler, I.; Khera, E.; Vance, S.; Kopp, A.; Qiu, Q.; Keating, T.A.; Abu-Yousif, A.O.; Sandal, T.; Legg, J.; Thompson, L.; et al. Increased Tumor Penetration of Single-Domain Antibody-Drug Conjugates Improves In Vivo Efficacy in Prostate Cancer Models. Cancer Res. 2020, 80, 1268–1278. [Google Scholar] [CrossRef]
- Ballot, E.; Ladoire, S.; Routy, B.; Truntzer, C.; Ghiringhelli, F. Tumor Infiltrating Lymphocytes Signature as a New Pan-Cancer Predictive Biomarker of Anti PD-1/PD-L1 Efficacy. Cancers 2020, 12, 2418. [Google Scholar] [CrossRef]
- Melake, M.J.; Smith, H.G.; Mansfield, D.; Davies, E.; Dillon, M.; Wilkins, A.C.; Patin, E.C.; Pedersen, M.; Buus, R.; Miah, A.B.; et al. 4-1BB is a target for immunotherapy in patients with undifferentiated pleomorphic sarcoma. bioRxiv 2020, 11, 197293. [Google Scholar]


{kind=link}
{kind=link}
| Target | Name of IMP | Reference |
|---|---|---|
| CD27 | Varlilumab | [31] |
| CD27 | MK-5890 | [32] |
| OX40 | PF-04518600 | [33] |
| OX40 | MEDI0562 | [34] |
| OX40 | MEDI6469 | [35] |
| OX40 | GSK3174998 | [36] |
| OX40 | MOXR0916 | [37] |
| OX40 | INCAGN01949 | [38] |
| OX40 | BMS-986178 | [39] |
| GITR | TRX518 | [40] |
| GITR | MK-4166 | [41] |
| GITR | MK-1248 | [42] |
| GITR | BMS-986156 | [43] |
| GITR | MEDI1873 | [44] |
| GITR | AMG288 | [45] |
| CD137 | Urelumab (BMS-663513) | [46,47] |
| CD137 | Utomilumab (PF-05082566) | [48] |
| Company | Target | Format | Development Stage | Reference |
|---|---|---|---|---|
| PRS343 | HER2 × CD137 | Anticalin binding protein fused to whole IgG | Phase I | NCT03650348 |
| GEN1046/BNT-311 | PD-L1 × CD137 | Fc silenced bispecific antibody | Phase I | NCT03917381 |
| RO7122290 | FAP × CD137L | Trimeric CD137L and Fab | EUDRACT 2017-003961-83 | |
| RO7227166 | CD19 × CD137L | Trimeric CD137L and Fab | Phase I | NCT04077723 |
| CB307 | PSMA × CD137 × HSA | VH only | Phase I | NCT04839991 |
| PRS-344 | PD-L1 × CD137 | Anticalin binding protein fused to whole IgG | Phase I | NCT03330561 |
| MCLA-145 | PD-L1 × CD137 | Full length IgG with two different heavy chains | Phase I | NCT03922204 |
| ES101/INBRX-105 | PD-L1 × CD137 | Single domain antibody with disabled Fc function | Phase I | NCT04009460 |
| NM21-1480 | PD-L1 × CD137 × HSA | 3 Fvs linked by linkers | Phase I | NCT04442126 |
| ABL503 | PD-L1 × CD137 | Anti-PDL1 mAb Fc silenced IgG fused with scFv of anti-CD137 | Phase I | NCT04762641 |
| FS222 | PD-L1 × CD137 | Tetravalent IgG with decreased FcγR binding | Phase I | NCT04740424 |
| FS120 | OX40 × CD137 | Tetravalent IgG with decreased FcγR binding | Phase I | NCT04648202 |
| GEN1042 | CD40 × CD137 | Fc silenced bispecific antibody | Phase I/IIa | NCT04083599 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, K. CD137 as an Attractive T Cell Co-Stimulatory Target in the TNFRSF for Immuno-Oncology Drug Development. Cancers 2021, 13, 2288. https://doi.org/10.3390/cancers13102288
Hashimoto K. CD137 as an Attractive T Cell Co-Stimulatory Target in the TNFRSF for Immuno-Oncology Drug Development. Cancers. 2021; 13(10):2288. https://doi.org/10.3390/cancers13102288
Chicago/Turabian StyleHashimoto, Kenji. 2021. "CD137 as an Attractive T Cell Co-Stimulatory Target in the TNFRSF for Immuno-Oncology Drug Development" Cancers 13, no. 10: 2288. https://doi.org/10.3390/cancers13102288
APA StyleHashimoto, K. (2021). CD137 as an Attractive T Cell Co-Stimulatory Target in the TNFRSF for Immuno-Oncology Drug Development. Cancers, 13(10), 2288. https://doi.org/10.3390/cancers13102288