
Recent Advances in Implantation-Based Genetic Modeling of Biliary Carcinogenesis in Mice



Abstract
:Simple Summary
Abstract

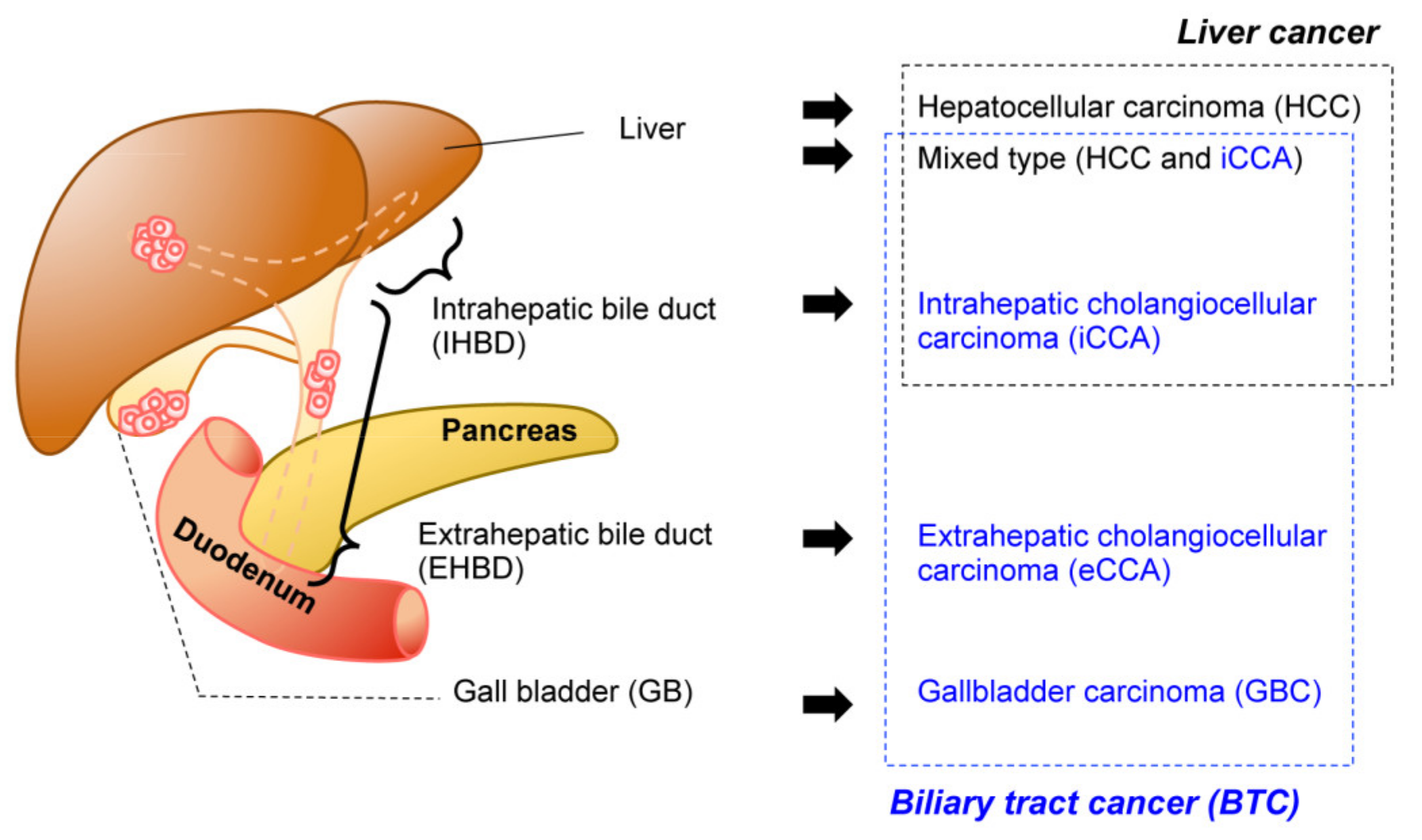
1. Introduction
2. Technical Overview of Genetic Mouse Models of BTC
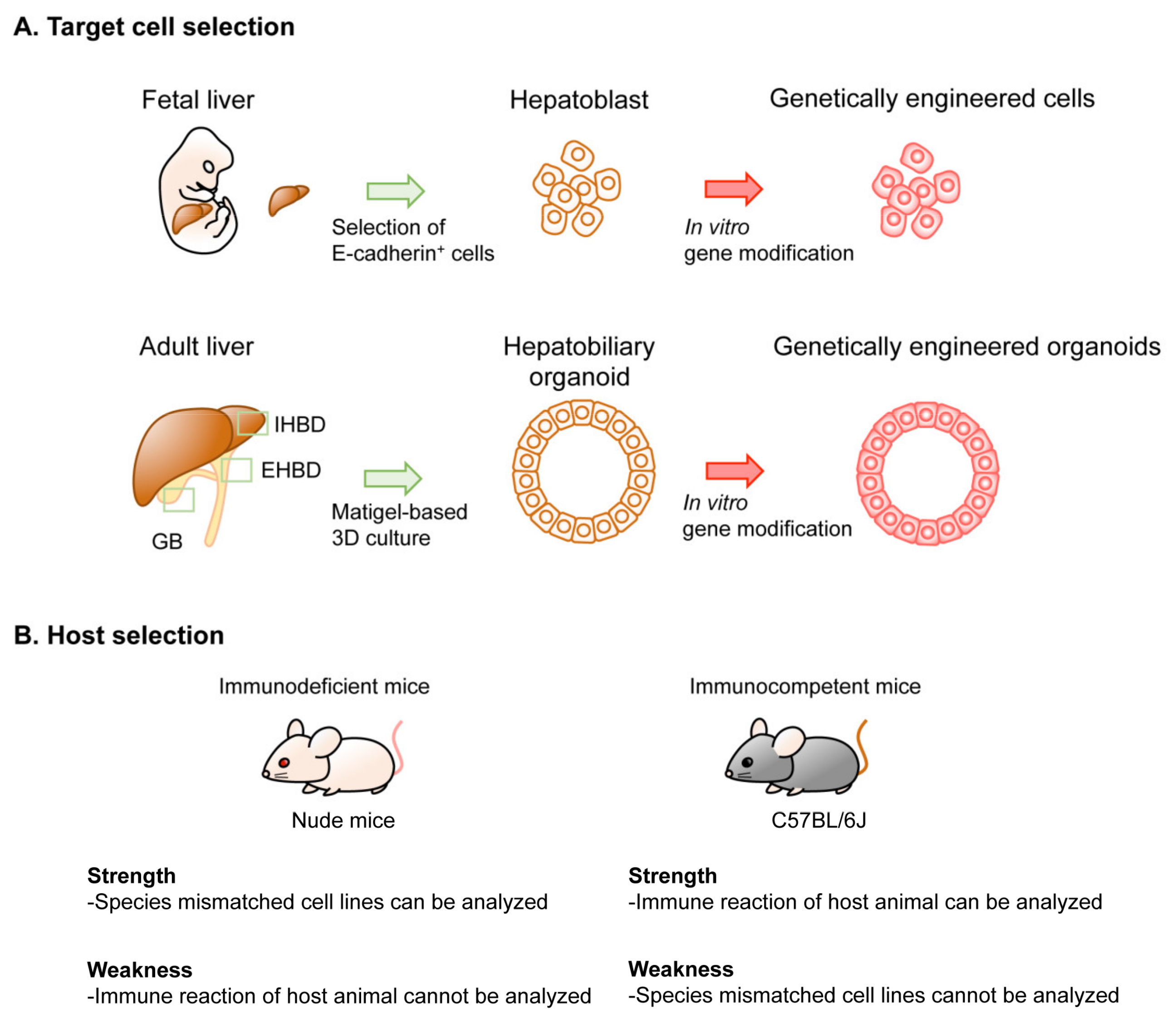
2.1. Target Cell Type
2.1.1. Specification by Cre Mice
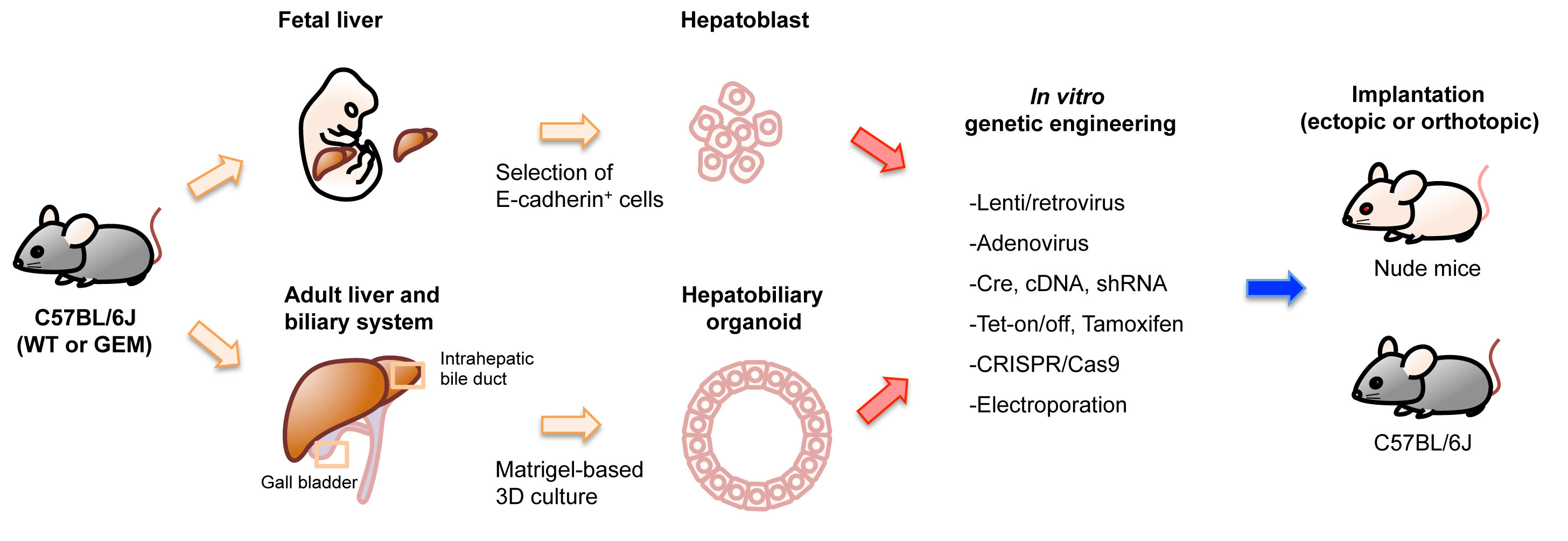
2.1.2. Specification by Physical Isolation Followed by Primary Culture
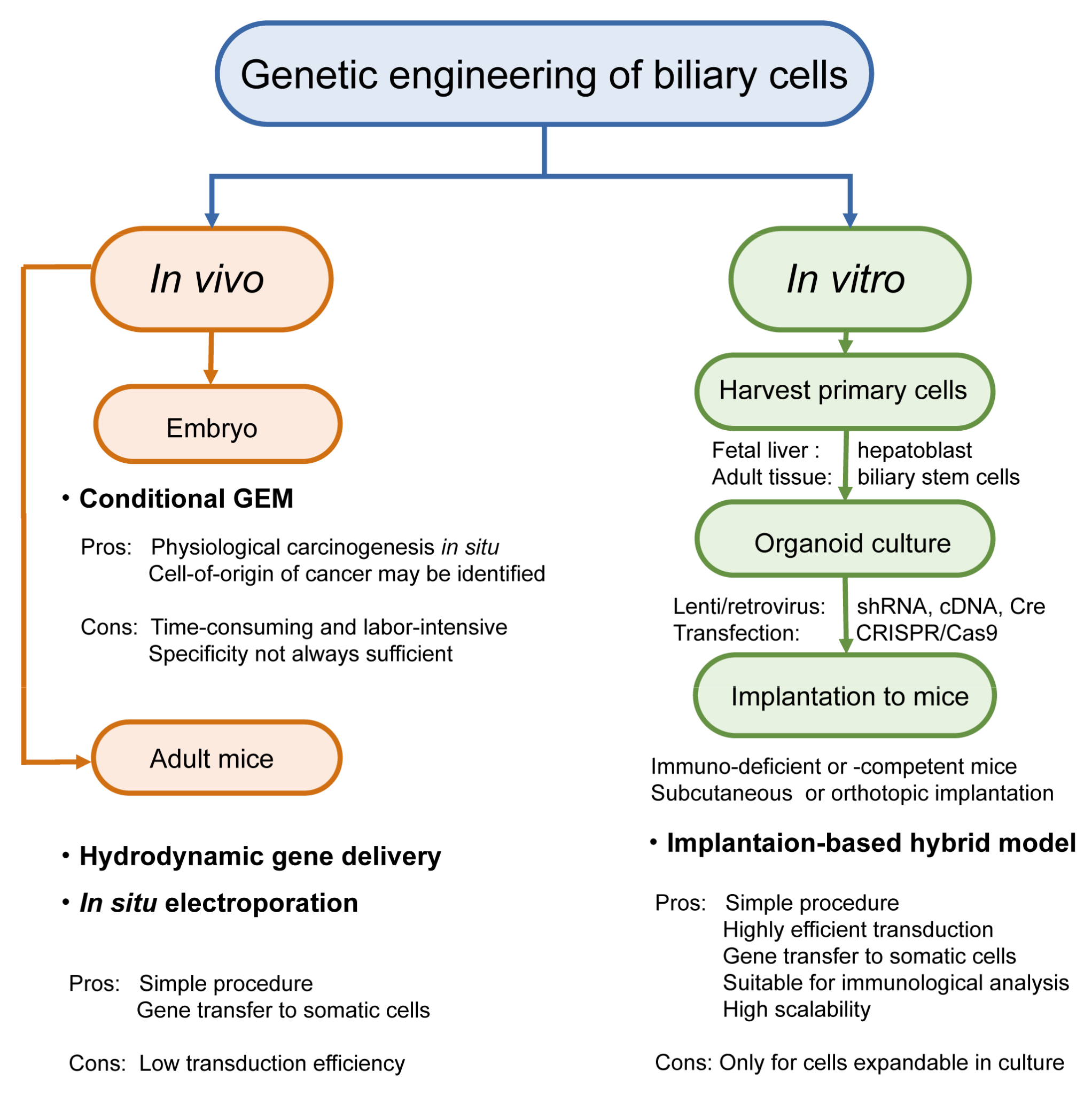
2.2. Genetic Engineering
2.2.1. In Vivo Genetic Engineering
Conditional GEM
Hydrodynamic Gene Delivery (HGD)
In Situ Electroporation
2.2.2. In Vitro Genetic Engineering
Viral Infection
CRISPR/Cas9
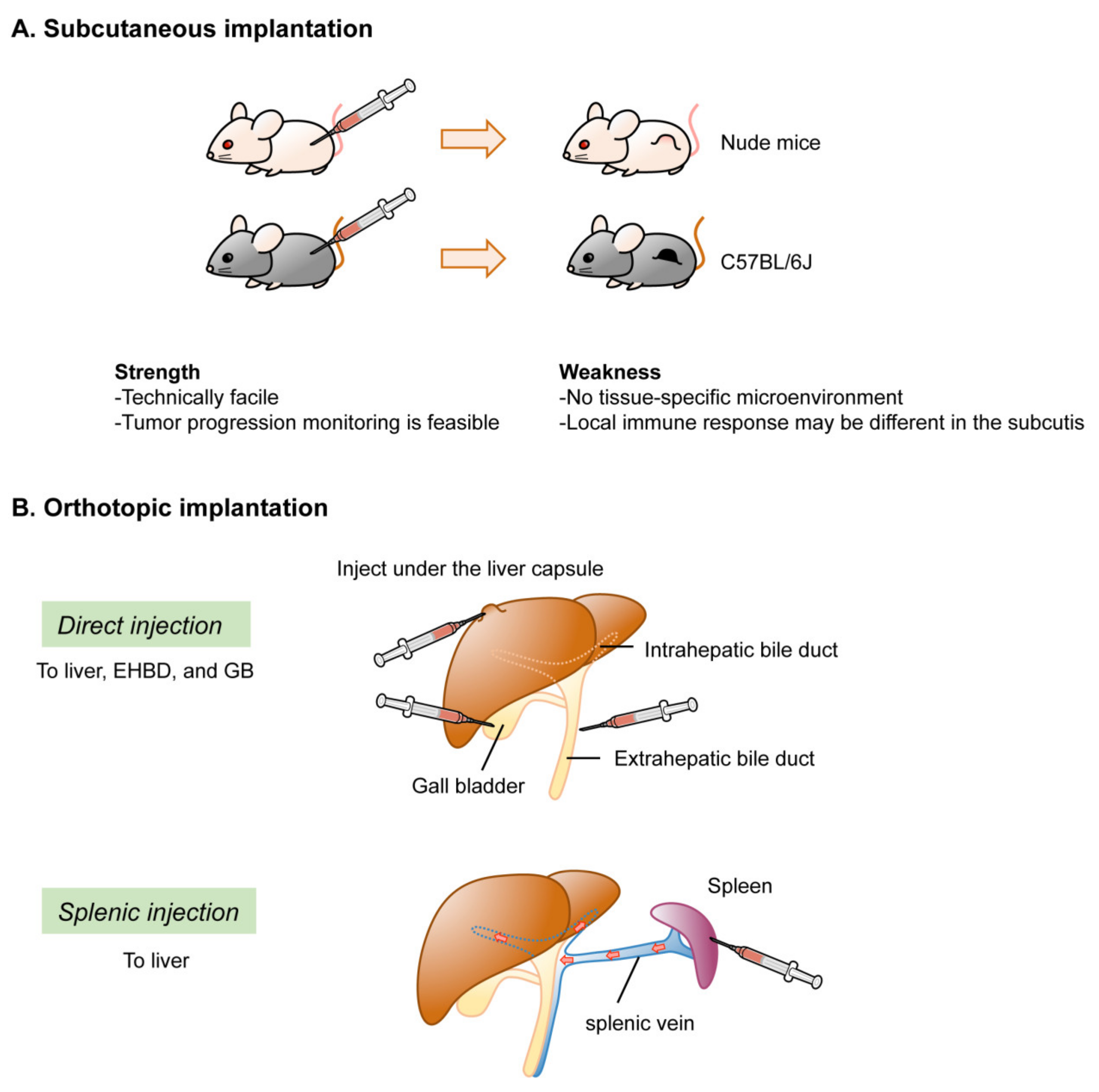
2.3. Recipient Mice
2.4. Location of Tumor Development
2.4.1. Ectopic Implantation
2.4.2. Orthotopic Implantation
3. In Vivo GEM Model of BTC
4. Implantation-Based Hybrid Mouse Model of BTC
4.1. Fetal Liver-Based Model of CCA and HCC
4.2. Organoid-Based Models of iCCA and eCCA
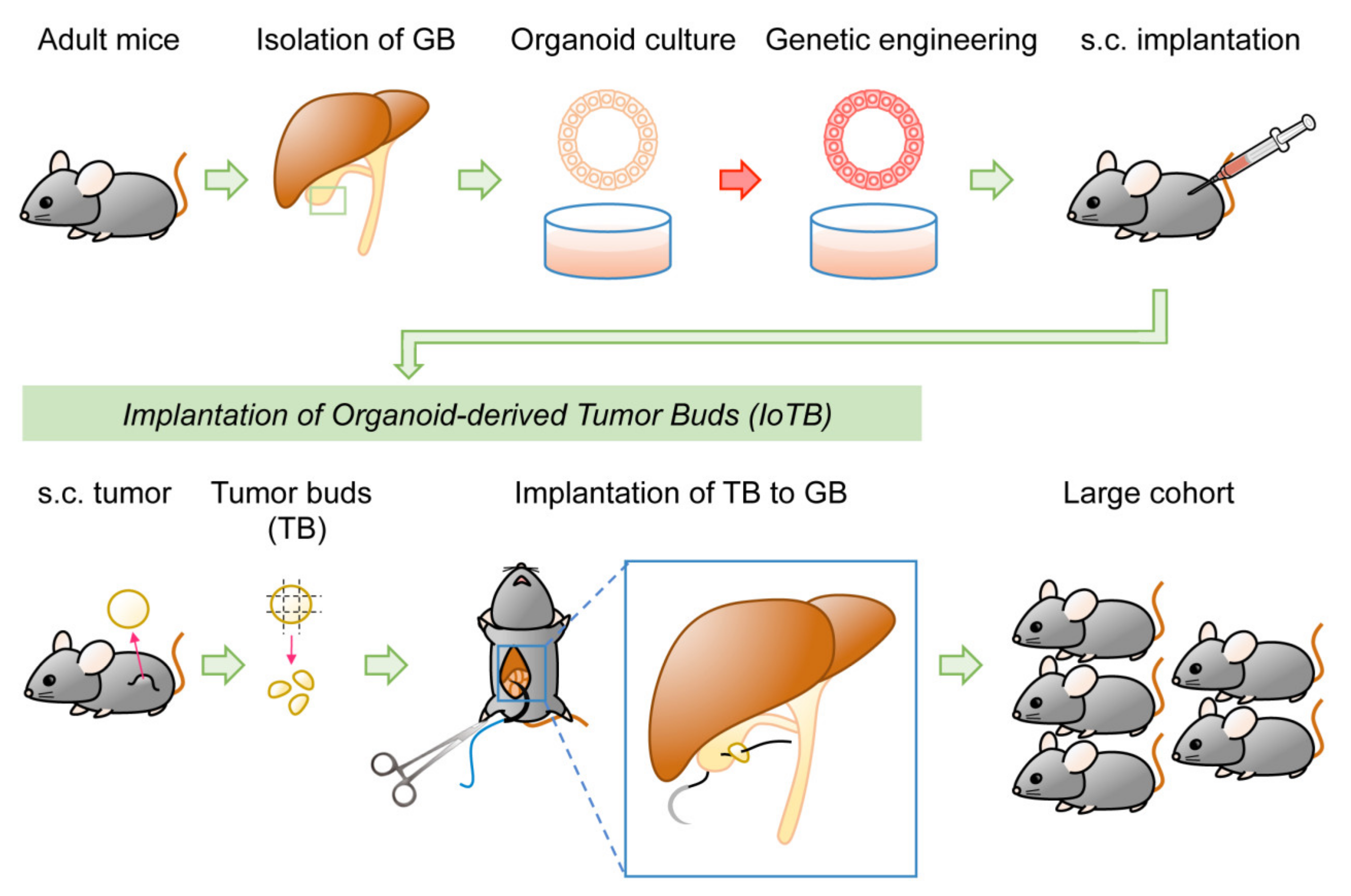
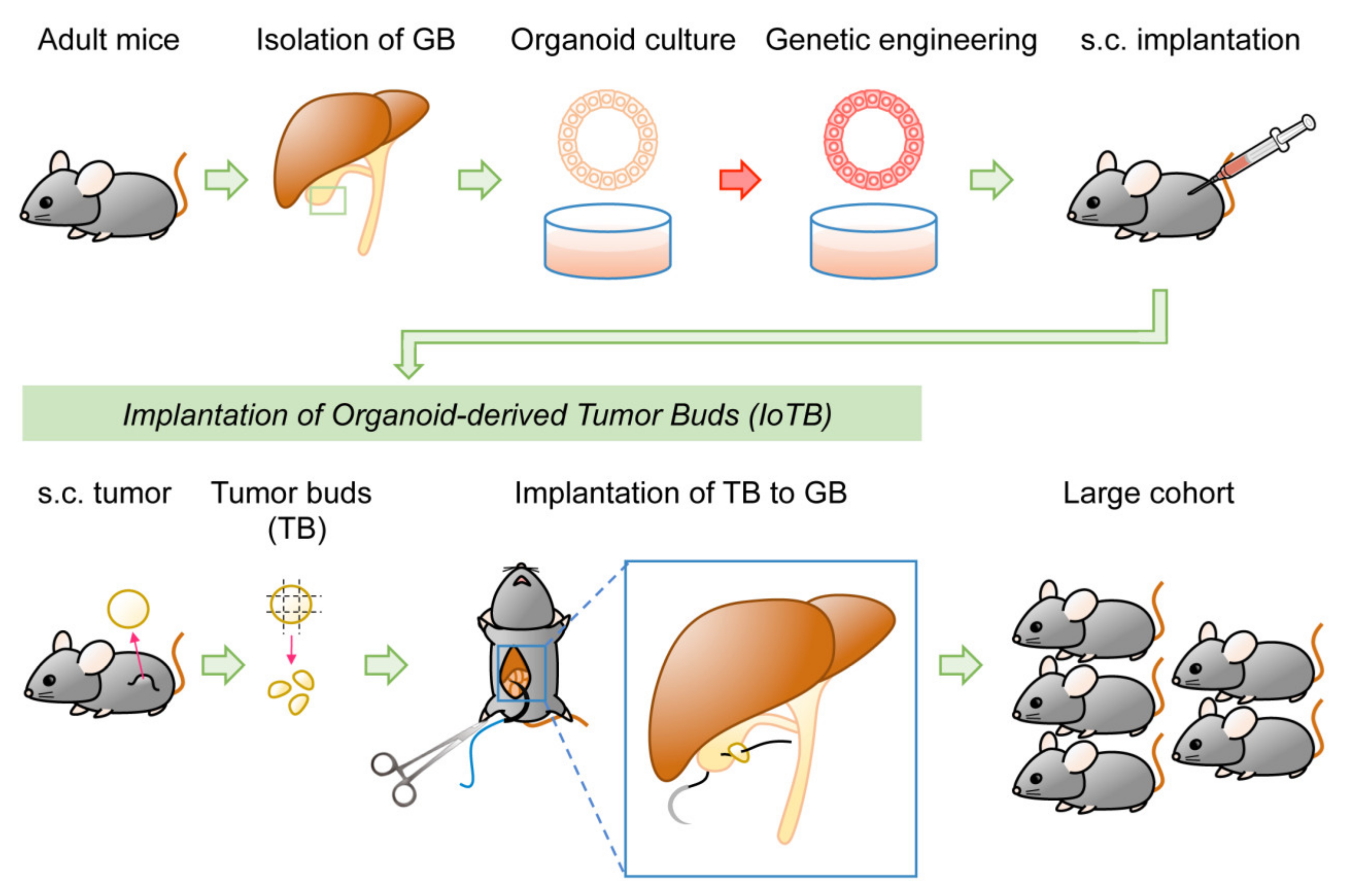
4.3. Organoid-Based Model of GBC
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef]
- Florio, A.A.; Ferlay, J.; Znaor, A.; Ruggieri, D.; Alvarez, C.S.; Laversanne, M.; Bray, F.; McGlynn, K.A.; Petrick, J.L. Global trends in intrahepatic and extrahepatic cholangiocarcinoma incidence from 1993 to 2012. Cancer 2020, 126, 2666–2678. [Google Scholar] [CrossRef] [PubMed]
- Ainechi, S.; Lee, H. Updates on Precancerous Lesions of the Biliary Tract: Biliary Precancerous Lesion. Arch. Pathol. Lab. Med. 2016, 140, 1285–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-On, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Massa, A.; Varamo, C.; Vita, F.; Tavolari, S.; Peraldo-Neia, C.; Brandi, G.; Rizzo, A.; Cavalloni, G.; Aglietta, M. Evolution of the Experimental Models of Cholangiocarcinoma. Cancers 2020, 12, 2308. [Google Scholar] [CrossRef]
- Leiting, J.L.; Murphy, S.J.; Bergquist, J.R.; Hernandez, M.C.; Ivanics, T.; Abdelrahman, A.M.; Yang, L.; Lynch, I.; Smadbeck, J.B.; Cleary, S.P.; et al. Biliary tract cancer patient-derived xenografts: Surgeon impact on individualized medicine. JHEP Rep. 2020, 2, 100068. [Google Scholar] [CrossRef] [Green Version]
- Waddell, S.H.; Boulter, L. Developing models of cholangiocarcinoma to close the translational gap in cancer research. Expert Opin. Investig. Drugs 2021, 30, 439–450. [Google Scholar] [CrossRef]
- Saborowski, A.; Wolff, K.; Spielberg, S.; Beer, B.; Hartleben, B.; Erlangga, Z.; Becker, D.; Dow, L.E.; Marhenke, S.; Woller, N.; et al. Murine Liver Organoids as a Genetically Flexible System to Study Liver Cancer In Vivo and In Vitro. Hepatol. Commun. 2019, 3, 423–436. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, M.; Yoshihara, Y.; Maru, Y.; Tetsuya, M.; Izumiya, M.; Imai, T.; Hippo, Y. Kras-driven heterotopic tumor development from hepatobiliary organoids. Carcinogenesis 2019, 40, 1142–1152. [Google Scholar] [CrossRef]
- Cristinziano, G.; Porru, M.; Lamberti, D.; Buglioni, S.; Rollo, F.; Amoreo, C.A.; Manni, I.; Giannarelli, D.; Cristofoletti, C.; Russo, G.; et al. FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- Kasuga, A.; Semba, T.; Sato, R.; Nobusue, H.; Sugihara, E.; Takaishi, H.; Kanai, T.; Saya, H.; Arima, Y. Oncogenic KRAS-expressing organoids with biliary epithelial stem cell properties give rise to biliary tract cancer in mice. Cancer Sci. 2021, 112, 1822–1838. [Google Scholar] [CrossRef]
- Nakagawa, H.; Suzuki, N.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Kinoshita, H.; Ihara, S.; Uchino, K.; Nishikawa, Y.; Ijichi, H.; et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl. Acad. Sci. USA 2017, 114, E3806–E3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlangga, Z.; Wolff, K.; Poth, T.; Peltzer, A.; Nahnsen, S.; Spielberg, S.; Timrott, K.; Woller, N.; Kuhnel, F.; Manns, M.P.; et al. Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer. Cancers 2019, 11, 1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Fushimi, K.; Yabuki, Y.; Maru, Y.; Hasegawa, S.; Matsuura, T.; Kurotaki, D.; Suzuki, A.; Kobayashi, N.; Yoneda, M.; et al. Precision modeling of gall bladder cancer patients in mice based on orthotopic implantation of organoid-derived tumor buds. Oncogenesis 2021, 10, 33. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhou, B. Strategies for site-specific recombination with high efficiency and precise spatiotemporal resolution. J. Biol. Chem. 2021, 100509. [Google Scholar] [CrossRef]
- McLellan, M.A.; Rosenthal, N.A.; Pinto, A.R. Cre-loxP-Mediated Recombination: General Principles and Experimental Considerations. Curr. Protoc. Mouse Biol. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Winters, I.P.; Murray, C.W.; Winslow, M.M. Towards quantitative and multiplexed in vivo functional cancer genomics. Nat. Rev. Genet. 2018, 19, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Di-Luoffo, M.; Pirenne, S.; Saandi, T.; Loriot, A.; Gerard, C.; Dauguet, N.; Manzano-Nunez, F.; Alves Souza Carvalhais, N.; Lamoline, F.; Cordi, S.; et al. A novel mouse model of cholangiocarcinoma uncovers a role for Tensin-4 in tumor progression. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Marsh, V.; Davies, E.J.; Williams, G.T.; Clarke, A.R. PTEN loss and KRAS activation cooperate in murine biliary tract malignancies. J. Pathol. 2013, 230, 165–173. [Google Scholar] [CrossRef]
- Ireland, H.; Kemp, R.; Houghton, C.; Howard, L.; Clarke, A.R.; Sansom, O.J.; Winton, D.J. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of beta-catenin. Gastroenterology 2004, 126, 1236–1246. [Google Scholar] [CrossRef]
- Kemp, R.; Ireland, H.; Clayton, E.; Houghton, C.; Howard, L.; Winton, D.J. Elimination of background recombination: Somatic induction of Cre by combined transcriptional regulation and hormone binding affinity. Nucleic Acids Res. 2004, 32, e92. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Fischer, S.; Serra, S.; Chetty, R. The use of Cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tulasi, D.Y.; Castaneda, D.M.; Wager, K.; Hogan, C.B.; Alcedo, K.P.; Raab, J.R.; Gracz, A.D. Sox9(EGFP) Defines Biliary Epithelial Heterogeneity Downstream of Yap Activity. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1437–1462. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Haber, P.K.; Sia, D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021, 3, 100226. [Google Scholar] [CrossRef] [PubMed]
- Newberry, E.P.; Xie, Y.; Lodeiro, C.; Solis, R.; Moritz, W.; Kennedy, S.; Barron, L.; Onufer, E.; Alpini, G.; Zhou, T.; et al. Hepatocyte and stellate cell deletion of liver fatty acid binding protein reveals distinct roles in fibrogenic injury. FASEB J. 2019, 33, 4610–4625. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grun, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Pepe-Mooney, B.J.; Dill, M.T.; Alemany, A.; Ordovas-Montanes, J.; Matsushita, Y.; Rao, A.; Sen, A.; Miyazaki, M.; Anakk, S.; Dawson, P.A.; et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 2019, 25, 23–38. [Google Scholar] [CrossRef]
- Wang, X.; Ni, C.; Jiang, N.; Wei, J.; Liang, J.; Zhao, B.; Lin, X. Generation of liver bipotential organoids with a small-molecule cocktail. J. Mol. Cell Biol. 2020, 12, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, D.; Rizvi, S.; Razumilava, N.; Bronk, S.F.; Davila, J.I.; Champion, M.D.; Borad, M.J.; Bezerra, J.A.; Chen, X.; Gores, G.J. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015, 61, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.M.; Frandsen, J.L.; Kirchhof, N.; McIvor, R.S.; Largaespada, D.A. Somatic integration of an oncogene-harboring Sleeping Beauty transposon models liver tumor development in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 17059–17064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evert, M.; Dombrowski, F.; Fan, B.; Ribback, S.; Chen, X.; Calvisi, D.F. On the role of notch1 and adult hepatocytes in murine intrahepatic cholangiocarcinoma development. Hepatology 2013, 58, 1857–1859. [Google Scholar] [CrossRef]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Deng, N.; Xu, Y.; Li, L.; Kuang, D.; Li, M.; Li, X.; Xu, Z.; Xiang, M.; Xu, C. Bmi1 drives the formation and development of intrahepatic cholangiocarcinoma independent of Ink4A/Arf repression. Pharmacol. Res. 2021, 164, 105365. [Google Scholar] [CrossRef]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar]
- Li, L.; Che, L.; Tharp, K.M.; Park, H.M.; Pilo, M.G.; Cao, D.; Cigliano, A.; Latte, G.; Xu, Z.; Ribback, S.; et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016, 63, 1900–1913. [Google Scholar] [CrossRef] [Green Version]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, H.; Peters, M.; Ding, N.; Ribback, S.; Utpatel, K.; Cigliano, A.; Dombrowski, F.; Xu, M.; Chen, X.; et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J. Hepatol. 2019, 71, 742–752. [Google Scholar] [CrossRef]
- Yamamoto, M.; Xin, B.; Watanabe, K.; Ooshio, T.; Fujii, K.; Chen, X.; Okada, Y.; Abe, H.; Taguchi, Y.; Miyokawa, N.; et al. Oncogenic Determination of a Broad Spectrum of Phenotypes of Hepatocyte-Derived Mouse Liver Tumors. Am. J. Pathol. 2017, 187, 2711–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Shin, B.C.; Fujikura, K.; Matsuzaki, T.; Takata, K. Direct gene transfer into rat liver cells by in vivo electroporation. FEBS Lett. 1998, 425, 436–440. [Google Scholar] [CrossRef] [Green Version]
- Gürlevik, E.; Fleischmann-Mundt, B.; Armbrecht, N.; Longerich, T.; Woller, N.; Kloos, A.; Hoffmann, D.; Schambach, A.; Wirth, T.C.; Manns, M.P.; et al. Adjuvant gemcitabine therapy improves survival in a locally induced, R0-resectable model of metastatic intrahepatic cholangiocarcinoma. Hepatology 2013, 58, 1031–1041. [Google Scholar] [CrossRef]
- Kendre, G.; Marhenke, S.; Lorz, G.; Becker, D.; Reineke-Plaass, T.; Poth, T.; Murugesan, K.; Kuhnel, F.; Woller, N.; Wirtz, R.M.; et al. The co-mutational spectrum determines the therapeutic response in murine FGFR2 fusion—Driven cholangiocarcinoma. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Dore, G.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Onuma, K.; Ochiai, M.; Orihashi, K.; Takahashi, M.; Imai, T.; Nakagama, H.; Hippo, Y. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11127–11132. [Google Scholar] [CrossRef] [Green Version]
- Maru, Y.; Onuma, K.; Ochiai, M.; Imai, T.; Hippo, Y. Shortcuts to intestinal carcinogenesis by genetic engineering in organoids. Cancer Sci. 2019, 110, 858–866. [Google Scholar] [CrossRef]
- Maru, Y.; Orihashi, K.; Hippo, Y. Lentivirus-Based Stable Gene Delivery into Intestinal Organoids. Methods Mol. Biol. 2016, 1422, 13–21. [Google Scholar]
- O’Doherty, U.; Swiggard, W.J.; Malim, M.H. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 2000, 74, 10074–10080. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Rubinson, D.A.; Dillon, C.P.; Kwiatkowski, A.V.; Sievers, C.; Yang, L.; Kopinja, J.; Rooney, D.L.; Zhang, M.; Ihrig, M.M.; McManus, M.T.; et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 2003, 33, 401–406. [Google Scholar] [CrossRef]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.O.; Wilcox, K.W. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J. Mol. Biol. 1970, 51, 379–391. [Google Scholar] [CrossRef]
- Kelly, T.J., Jr.; Smith, H.O. A restriction enzyme from Hemophilus influenzae. II. J. Mol. Biol. 1970, 51, 393–409. [Google Scholar] [CrossRef]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Danna, K.; Nathans, D. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc. Natl. Acad. Sci. USA 1971, 68, 2913–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonte, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeggo, P.A. DNA breakage and repair. Adv. Genet. 1998, 38, 185–218. [Google Scholar] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Clevers, H. CRISPR/Cas 9 genome editing and its applications in organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G257–G265. [Google Scholar] [CrossRef] [PubMed]
- Artegiani, B.; Hendriks, D.; Beumer, J.; Kok, R.; Zheng, X.; Joore, I.; Chuva de Sousa Lopes, S.; van Zon, J.; Tans, S.; Clevers, H. Fast and efficient generation of knock-in human organoids using homology-independent CRISPR-Cas9 precision genome editing. Nat. Cell Biol. 2020, 22, 321–331. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Balmain, A.; Harris, C.C. Carcinogenesis in mouse and human cells: Parallels and paradoxes. Carcinogenesis 2000, 21, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T.; Maru, Y.; Izumiya, M.; Hoshi, D.; Kato, S.; Ochiai, M.; Hori, M.; Yamamoto, S.; Tatsuno, K.; Imai, T.; et al. Organoid-based ex vivo reconstitution of Kras-driven pancreatic ductal carcinogenesis. Carcinogenesis 2020, 41, 490–501. [Google Scholar] [CrossRef]
- Stripecke, R.; Munz, C.; Schuringa, J.J.; Bissig, K.D.; Soper, B.; Meeham, T.; Yao, L.C.; Di Santo, J.P.; Brehm, M.; Rodriguez, E.; et al. Innovations, challenges, and minimal information for standardization of humanized mice. EMBO Mol. Med. 2020, 12, e8662. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Sojoodi, M.; Taylor, M.S.; Ghoshal, S.; Razavi, A.A.; Graham-O’Regan, K.A.; Bardeesy, N.; Ferrone, C.R.; Lanuti, M.; Caravan, P.; et al. Orthotopic and heterotopic murine models of pancreatic cancer and their different responses to FOLFIRINOX chemotherapy. Dis. Model. Mech. 2018, 11, dmm034793. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Izhak, L.; Ambrosino, E.; Kato, S.; Parish, S.T.; O’Konek, J.J.; Weber, H.; Xia, Z.; Venzon, D.; Berzofsky, J.A.; Terabe, M. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res. 2013, 73, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Saborowski, A.; Saborowski, M.; Davare, M.A.; Druker, B.J.; Klimstra, D.S.; Lowe, S.W. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc. Natl. Acad. Sci. USA 2013, 110, 19513–19518. [Google Scholar] [CrossRef] [Green Version]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zender, L.; Xue, W.; Cordon-Cardo, C.; Hannon, G.J.; Lucito, R.; Powers, S.; Flemming, P.; Spector, M.S.; Lowe, S.W. Generation and analysis of genetically defined liver carcinomas derived from bipotential liver progenitors. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Egberts, J.H.; Schniewind, B.; Schafmayer, C.; Kruse, M.L.; Sipos, B.; Fandrich, F.; Kalthoff, H.; Tepel, J. Establishment of a novel orthotopic xenograft model of human gallbladder carcinoma. Clin. Exp. Metastasis 2007, 24, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Kawamata, H.; Fujimori, T.; Kuroda, Y. A MEK inhibitor (U0126) prolongs survival in nude mice bearing human gallbladder cancer cells with K-ras mutation: Analysis in a novel orthotopic inoculation model. Int. J. Oncol. 2003, 23, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kobayashi, S.; Qiao, W.; Li, C.; Xiao, C.; Radaeva, S.; Stiles, B.; Wang, R.H.; Ohara, N.; Yoshino, T.; et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J. Clin. Investig. 2006, 116, 1843–1852. [Google Scholar] [CrossRef]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef] [PubMed]
- Galicia, V.A.; He, L.; Dang, H.; Kanel, G.; Vendryes, C.; French, B.A.; Zeng, N.; Bayan, J.A.; Ding, W.; Wang, K.S.; et al. Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 2010, 139, 2170–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.A.; Alexander, W.B.; Guo, B.; Kato, Y.; Patra, K.; O’Dell, M.R.; McCall, M.N.; Whitney-Miller, C.L.; Bardeesy, N.; Hezel, A.F. Kras and Tp53 Mutations Cause Cholangiocyte- and Hepatocyte-Derived Cholangiocarcinoma. Cancer Res. 2018, 78, 4445–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenoue, T.; Terakado, Y.; Zhu, C.; Liu, X.; Ohsugi, T.; Matsubara, D.; Fujii, T.; Kakuta, S.; Kubo, S.; Shibata, T.; et al. Establishment and analysis of a novel mouse line carrying a conditional knockin allele of a cancer-specific FBXW7 mutation. Sci. Rep. 2018, 8, 2021. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Lee, J.H.; Kim, T.S.; Kim, T.H.; Park, H.D.; Byun, J.S.; Kim, M.C.; Jeong, W.I.; Calvisi, D.F.; Kim, J.M.; et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.K.; Fang, Z.; Jiang, T.Y.; Wan, Z.H.; Pan, Y.F.; Ma, Y.H.; Shi, Y.Y.; Tan, Y.X.; Dong, L.W.; Zhang, Y.J.; et al. Combination of Kras activation and PTEN deletion contributes to murine hepatopancreatic ductal malignancy. Cancer Lett. 2018, 421, 161–169. [Google Scholar] [CrossRef]
- O’Dell, M.R.; Huang, J.L.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sorensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, A.L.; Xu, Y.; Zhao, A.; Ray, K.C.; Gu, G. A CK19(CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis 2008, 46, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.C.; Wang, J.; Zhou, Y.; Xu, K. Kras(G12D) upregulates Notch signaling to induce gallbladder tumorigenesis in mice. Oncoscience 2017, 4, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Kiguchi, K.; Bol, D.; Carbajal, S.; Beltran, L.; Moats, S.; Chan, K.; Jorcano, J.; DiGiovanni, J. Constitutive expression of erbB2 in epidermis of transgenic mice results in epidermal hyperproliferation and spontaneous skin tumor development. Oncogene 2000, 19, 4243–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltran, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar]
- Sato, T.; Morita, M.; Tanaka, R.; Inoue, Y.; Nomura, M.; Sakamoto, Y.; Miura, K.; Ito, S.; Sato, I.; Tanaka, N.; et al. Ex vivo model of non-small cell lung cancer using mouse lung epithelial cells. Oncol. Lett. 2017, 14, 6863–6868. [Google Scholar] [CrossRef]
- Naruse, M.; Masui, R.; Ochiai, M.; Maru, Y.; Hippo, Y.; Imai, T. An organoid-based carcinogenesis model induced by in vitro chemical treatment. Carcinogenesis 2020, 41, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, H.K.; Orkin, S.H. The journey of developing hematopoietic stem cells. Development 2006, 133, 3733–3744. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.; Yoshimoto, M.; Takebe, T. Fetal liver hematopoiesis: From development to delivery. Stem Cell Res. Ther. 2021, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, M.; Hippo, Y.; Izumiya, M.; Watanabe, M.; Nakagama, H. Newly defined aberrant crypt foci as a marker for dysplasia in the rat colon. Cancer Sci. 2014, 105, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Driver Oncogenes | Genotype of Organoids * | Methods for Genetic Engineering ** | Host | Implantation | Ref. | |
|---|---|---|---|---|---|---|
| Oncogenes | TSGs | |||||
| HCC (Hepatocellular Carcinoma) from Liver Organoid | ||||||
| cMyc | WT | cDNA (R) | shRNA (R); Trp53 and CRISPR/Cas9 (T); Apc | C57BL/6J | liver | [18] |
| iCCA (Intrahepatic Cholangiocelular Carcinoma) from Liver Organoid | ||||||
| KrasG12D | KrasLSL-G12D/+ | Cre (L) | shRNA (L): Cdkn2a and/or Pten, Trp53, Apc | Nude | s.c. | [19] |
| KrasLSL-G12D/+; Trp53flox/flox | N.T. | |||||
| Pik3caH1047R | Pik3caH1047R | shRNA (L): Cdkn2a, Pten, | ||||
| Rosa26- Pik3caH1047R; Trp53flox/flox | N.T. | |||||
| FGFR2-AHCYL1 | WT | cDNA(R) | shRNA (L): Cdkn2a and/or Pten | |||
| KrasG12D | KrasLSL-G12D/+; Trp53flox/flox (outbred) | Cre (R) | N.T. or shRNA (R): Pten | NSG | s.c. or liver | [18] |
| KrasLSL-G12D/+ | Cre (T) | CRISPR/Cas9 (T): Pten and Trp53 | C57BL/6J | liver | ||
| FGFR2-BICC1, -MGEA5, -TACC3 | Trp53−/− | cDNA (R) | N.T. | NOD-SCID | s.c. or liver | [20] |
| KRASG12V | Cdkn2a−/− | cDNA (R) | C57BL/6J | s.c., liver, or kidney | [21] | |
| eCCA (Extrahepatic Cholangiocelular Carcinoma) from CBD Organoid | ||||||
| KrasG12D | KrasLSL-G12D; Tgfbr2flox/flox; Cdh1flox/flox | Cre (L) | N.T. | Nude or C57BL/6J | s.c. | [22] |
| KRASG12V | Cdkn2a−/− | cDNA (R) | N.T. | C57BL/6J | s.c., liver, or kidney | [21] |
| GBC (Gallbladder Carcinoma) from GB Organoid | ||||||
| KrasG12D | KrasLSL-G12D | Cre (L) | shRNA (L): Cdkn2a, Pten | Nude | s.c. | [19] |
| KrasLSL-G12D/+; Trp53flox/flox | N.T. | |||||
| KRASG12V | Cdkn2a−/− | cDNA (R) | N.T. | C57BL/6J | s.c., liver, or kidney | [21] |
| KrasG12D | WT | Cre (T) | CRISPR/Cas9 (T): Pten and Trp53 | C57BL/6J | s.c. or GB | [23] |
| ERBB2S310F or V777L | cDNA (R) | CRISPR/Cas9 (T): Trp53 | NSG | s.c. | ||
| Rosa26- Pik3caH1047R; Trp53flox/flox | Cre (L) | N.T. | Nude | s.c. | [24] | |
| KrasG12D | KrasLSL-G12D | CRISPR/Cas9 (T): Trp53, p19Arf, Smad4 | C57BL/6J | GB via s.c. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izumiya, M.; Kato, S.; Hippo, Y. Recent Advances in Implantation-Based Genetic Modeling of Biliary Carcinogenesis in Mice. Cancers 2021, 13, 2292. https://doi.org/10.3390/cancers13102292
Izumiya M, Kato S, Hippo Y. Recent Advances in Implantation-Based Genetic Modeling of Biliary Carcinogenesis in Mice. Cancers. 2021; 13(10):2292. https://doi.org/10.3390/cancers13102292
Chicago/Turabian StyleIzumiya, Masashi, Shingo Kato, and Yoshitaka Hippo. 2021. "Recent Advances in Implantation-Based Genetic Modeling of Biliary Carcinogenesis in Mice" Cancers 13, no. 10: 2292. https://doi.org/10.3390/cancers13102292
APA StyleIzumiya, M., Kato, S., & Hippo, Y. (2021). Recent Advances in Implantation-Based Genetic Modeling of Biliary Carcinogenesis in Mice. Cancers, 13(10), 2292. https://doi.org/10.3390/cancers13102292