
Tumor Extrinsic Factors Mediate Primary T-DM1 Resistance in HER2-Positive Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
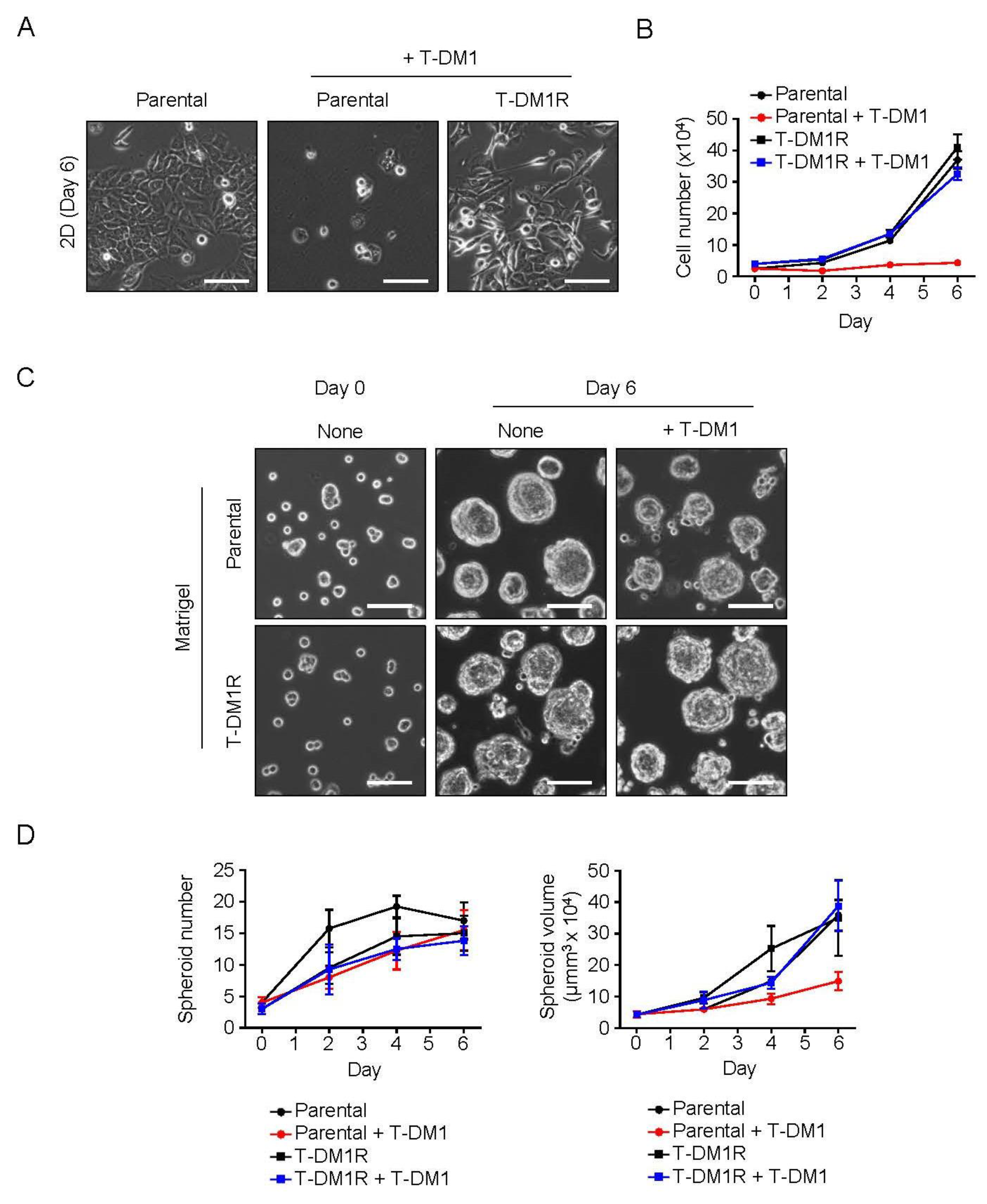
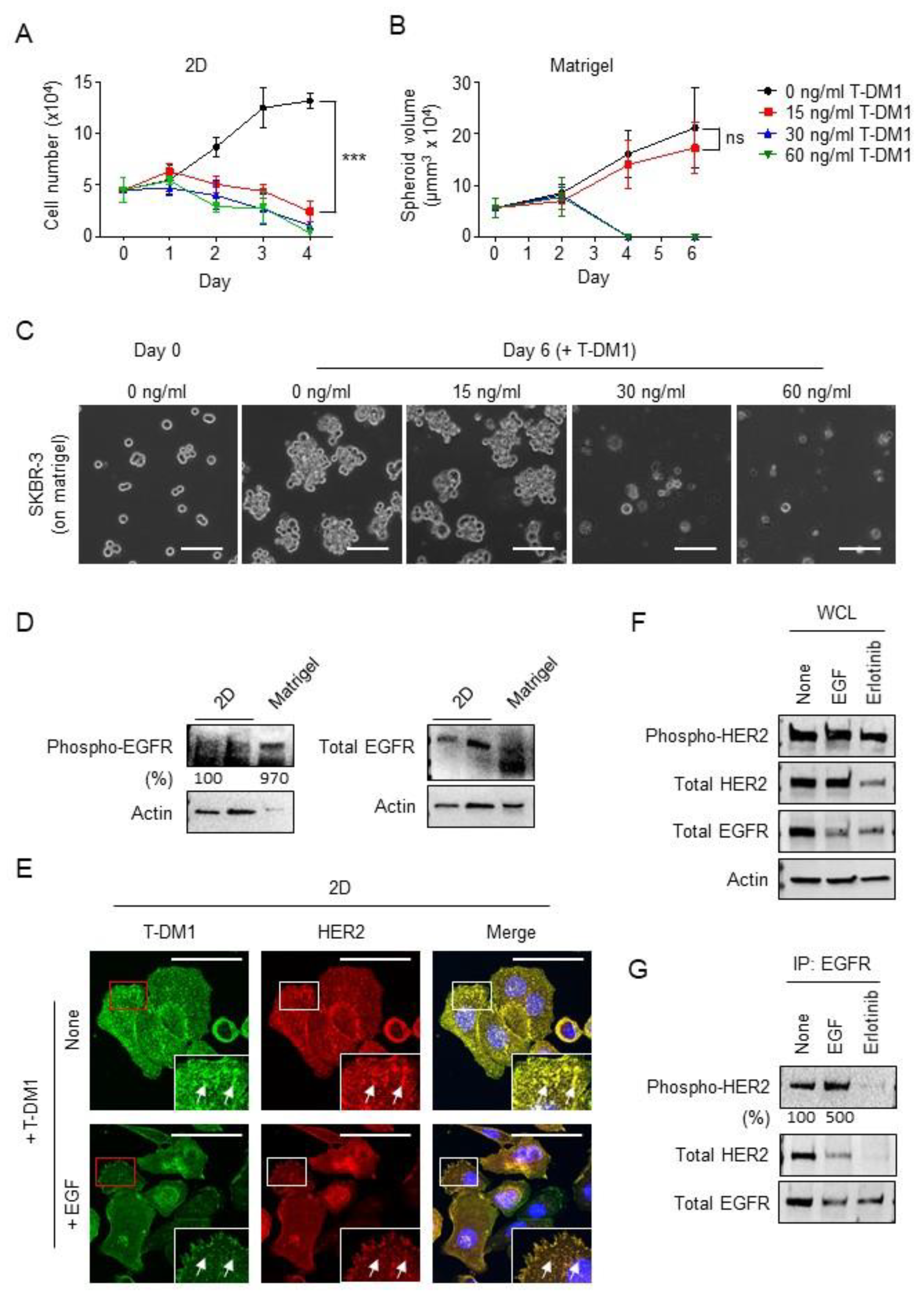
2.1. Spheroid Formation of the Parental JIMT1 Cells on Matrigel in the Presence of T-DM1
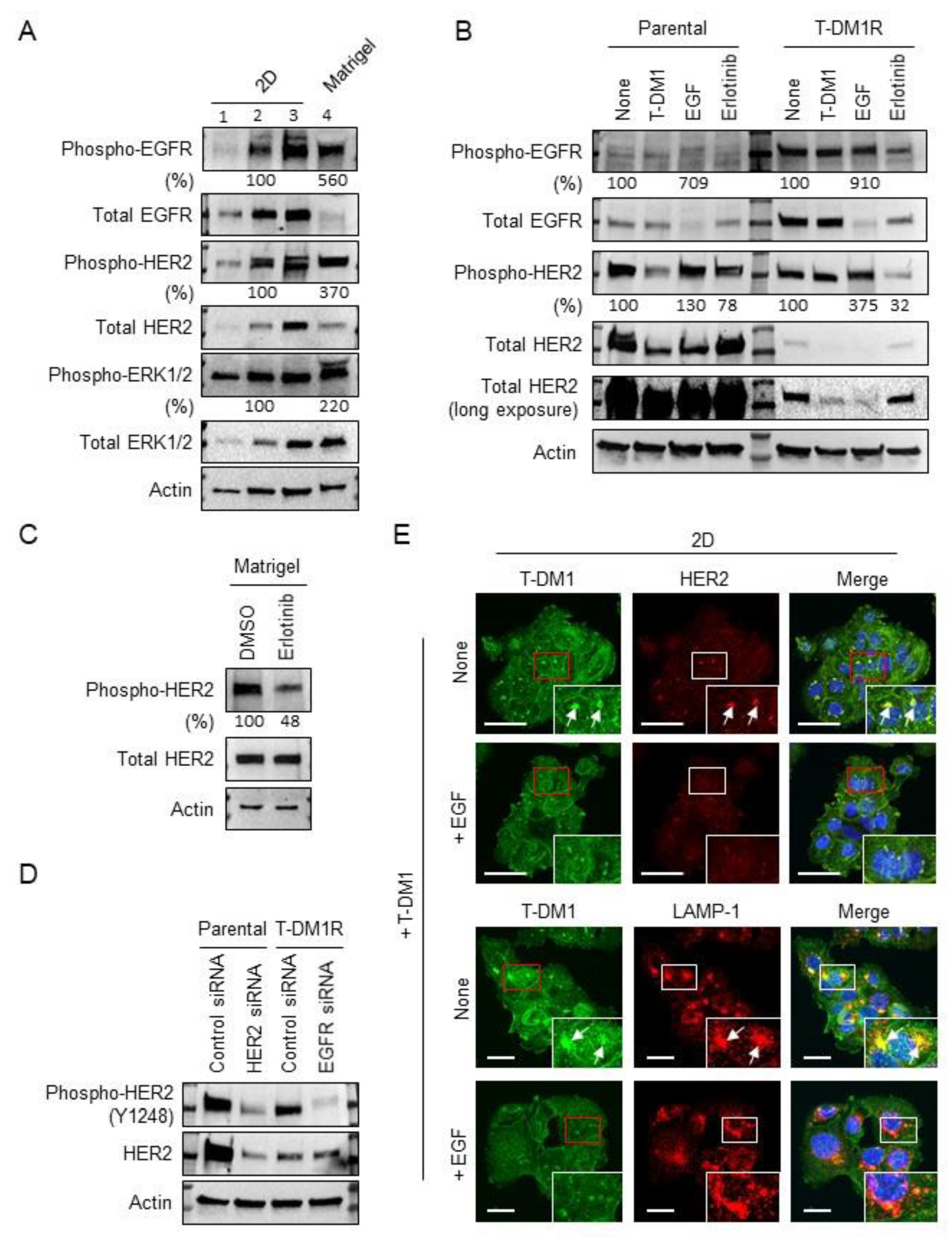
2.2. EGFR Activation and HER2 Phosphorylation of JIMT1 Cell Spheroids on Matrigel
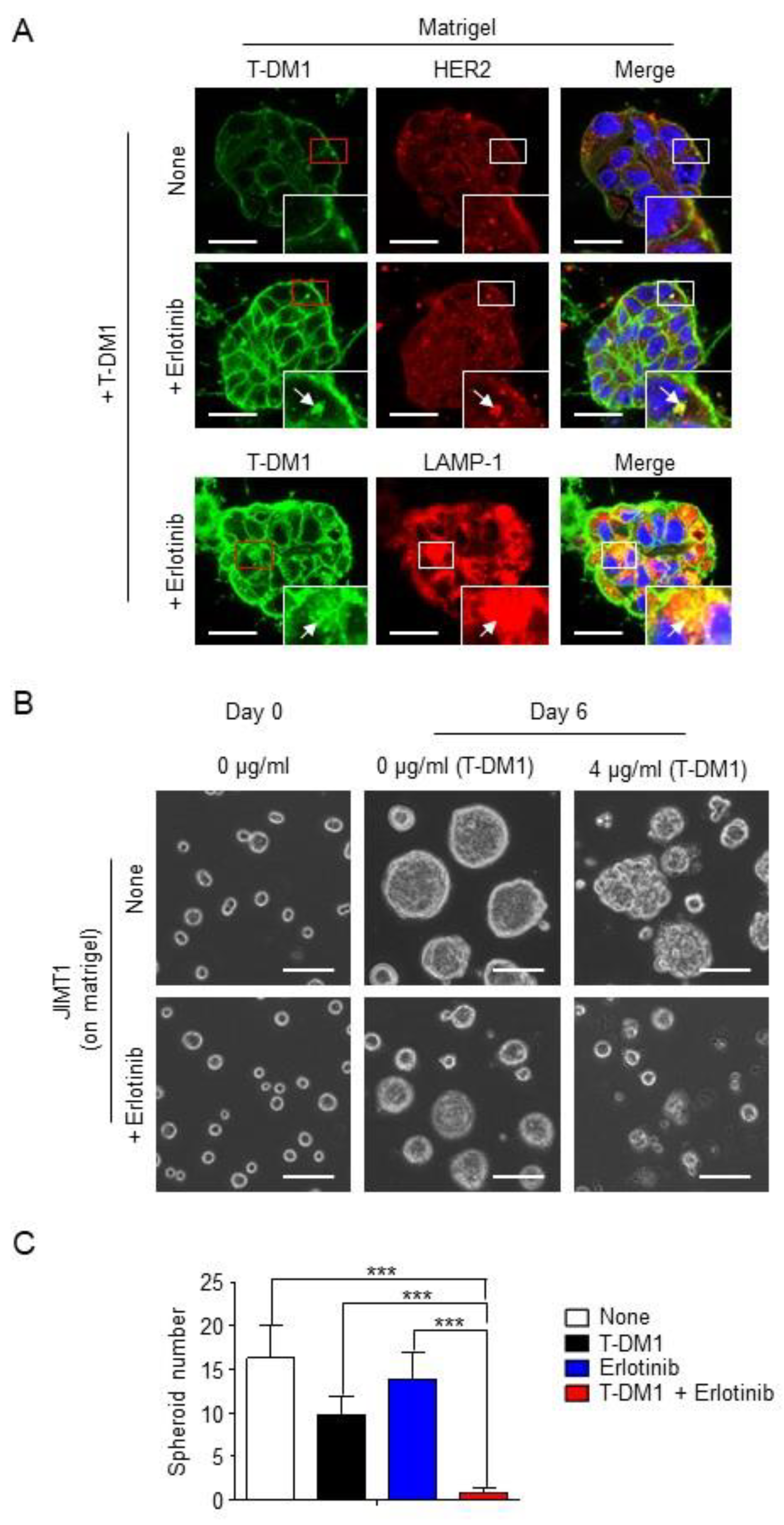
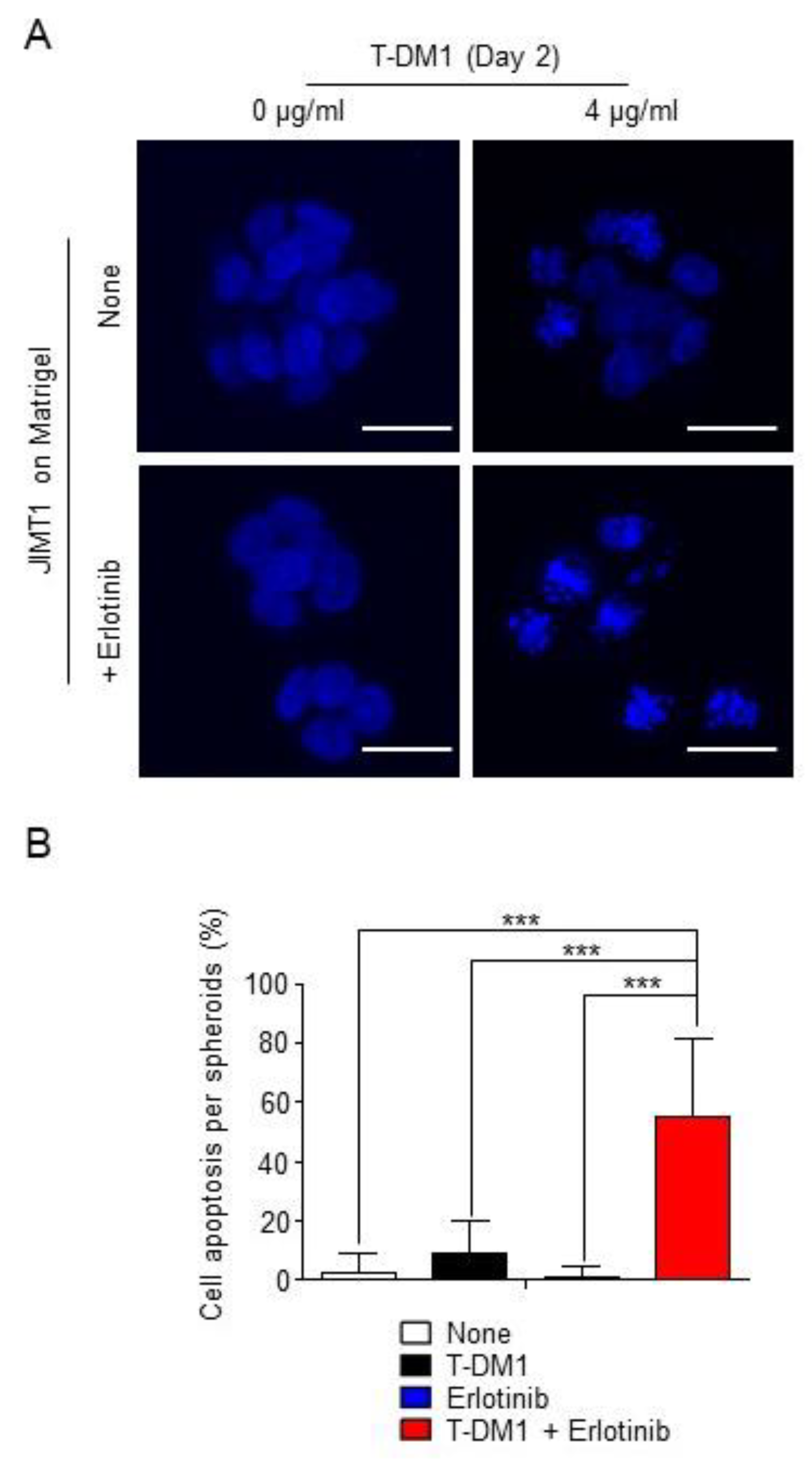
2.3. Combination of T-DM1 with Erlotinib Kills JIMT1-Spheroids on Matrigel
2.4. SKBR-3 Cells Are Less Sensitive to T-DM1 and Capable of Developing Spheroids on the Matrigel
3. Discussion
4. Materials and Methods
4.1. Cells, Therapeutic Drugs and Matrigel/ECM Gel
4.2. Cell Growth and Matrigel Assays
4.3. Immunofluorescent Staining
4.4. Western Blotting and Immunoprecipitation
4.5. siRNA Transfection
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Sarmento-Ribeiro, A.B.; Scorilas, A.; Gonçalves, A.C.; Efferth, T.; Trougakos, I.P. The emergence of drug resistance to tar-geted cancer therapies: Clinical evidence. Drug Resist. Updates 2019, 47, 100646. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; ELZarrad, K.M.; Hirsch, D.S.; Wu, J.W. Antibody-drug conjugates as therapeutic agents in oncology: Overview and perspective. Front. Anti-Cancer Drug Discov. 2013, 51, 139–189. [Google Scholar]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.H.; Temin, S.; Kirshner, J.J.; Chandarlapaty, S.; Crews, J.R.; Davidson, N.E.; Esteva, F.J.; Gonzalez-Angulo, A.M.; Krop, I.; Levinson, J.; et al. Systemic Therapy for Patients With Advanced Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2014, 32, 2078–2099. [Google Scholar] [CrossRef]
- Erickson, H.K.; Phillips, G.D.L.; Leipold, D.D.; Provenzano, C.A.; Mai, E.; Johnson, H.A.; Gunter, B.; Audette, C.A.; Gupta, M.; Pinkas, J.; et al. The Effect of Different Linkers on Target Cell Catabolism and Pharmacokinetics/Pharmacodynamics of Trastuzumab Maytansinoid Conjugates. Mol. Cancer Ther. 2012, 11, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Boyraz, B.; Sendur, M.A.; Aksoy, S.; Babacan, T.; Roach, E.C.; Kizilarslanoglu, M.C.; Petekkaya, I.; Altundag, K. Trastuzumab emtansine (T-DM1) for HER2-positive breast cancer. Curr. Med. Res. Opin. 2013, 29, 405–414. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Shen, Y.; Youssef, L.A.; Mohan, N.; Wu, J.W. T-DM1-resistant cells gain high invasive activity via EGFR and integ-rin cooperated pathway. Mabs 2018, 10, 1003–1017. [Google Scholar]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.; Kapanen, A.I.; Junttila, T.; Raheem, O.; Grenman, S.; Elo, J.; Elenius, K.; Isola, J. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol. Cancer Ther. 2004, 3, 1585–1592. [Google Scholar] [PubMed]
- Köninki, K.; Barok, M.; Tanner, M.; Staff, S.; Pitkänen, J.; Hemmilä, P.; Ilvesaro, J.; Isola, J. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010, 294, 211–219. [Google Scholar] [CrossRef]
- Kleinman, H.K.; McGarvey, M.L.; Liotta, L.A.; Robey, P.G.; Tryggvason, K.; Martin, G.R. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry 1982, 21, 6188–6193. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, H.K.; McGarvey, M.L.; Hassell, J.R.; Star, V.L.; Cannon, F.B.; Laurie, G.W.; Martin, G.R. Basement membrane complexes with biological activity. Biochemistry 1986, 25, 312–318. [Google Scholar] [CrossRef]
- Vukicevic, S.; Kleinman, H.K.; Luyten, F.P.; Roberts, A.B.; Roche, N.S.; Reddi, A.H. Identification of multiple active growth factors in basement membrane matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp. Cell Res. 1992, 202, 1–8. [Google Scholar] [CrossRef]
- McGuire, P.G.; Seeds, N.W. The interaction of plasminogen activator with a reconstituted basement membrane matrix and extracellular macromolecules produced by cultured epithelial cells. J. Cell. Biochem. 1989, 40, 215–227. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Yarden, Y. Ubiquitin ligase activity and tyro-sine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell. 1999, 4, 1029–1040. [Google Scholar] [CrossRef]
- Wu, W.J.; Tu, S.; Cerione, R.A. Activated Cdc42 Sequesters c-Cbl and Prevents EGF Receptor Degradation. Cell 2003, 114, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; González-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab Has Preferential Activity against Breast Cancers Driven by HER2 Homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endo, Y.; Wu, W.J. Tumor Extrinsic Factors Mediate Primary T-DM1 Resistance in HER2-Positive Breast Cancer Cells. Cancers 2021, 13, 2331. https://doi.org/10.3390/cancers13102331
Endo Y, Wu WJ. Tumor Extrinsic Factors Mediate Primary T-DM1 Resistance in HER2-Positive Breast Cancer Cells. Cancers. 2021; 13(10):2331. https://doi.org/10.3390/cancers13102331
Chicago/Turabian StyleEndo, Yukinori, and Wen Jin Wu. 2021. "Tumor Extrinsic Factors Mediate Primary T-DM1 Resistance in HER2-Positive Breast Cancer Cells" Cancers 13, no. 10: 2331. https://doi.org/10.3390/cancers13102331
APA StyleEndo, Y., & Wu, W. J. (2021). Tumor Extrinsic Factors Mediate Primary T-DM1 Resistance in HER2-Positive Breast Cancer Cells. Cancers, 13(10), 2331. https://doi.org/10.3390/cancers13102331