
Chondrosarcoma-from Molecular Pathology to Novel Therapies

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
:Simple Summary
Abstract
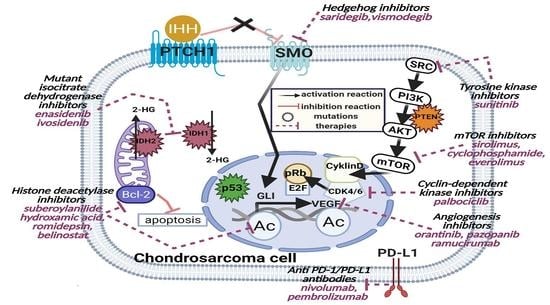
1. Introduction
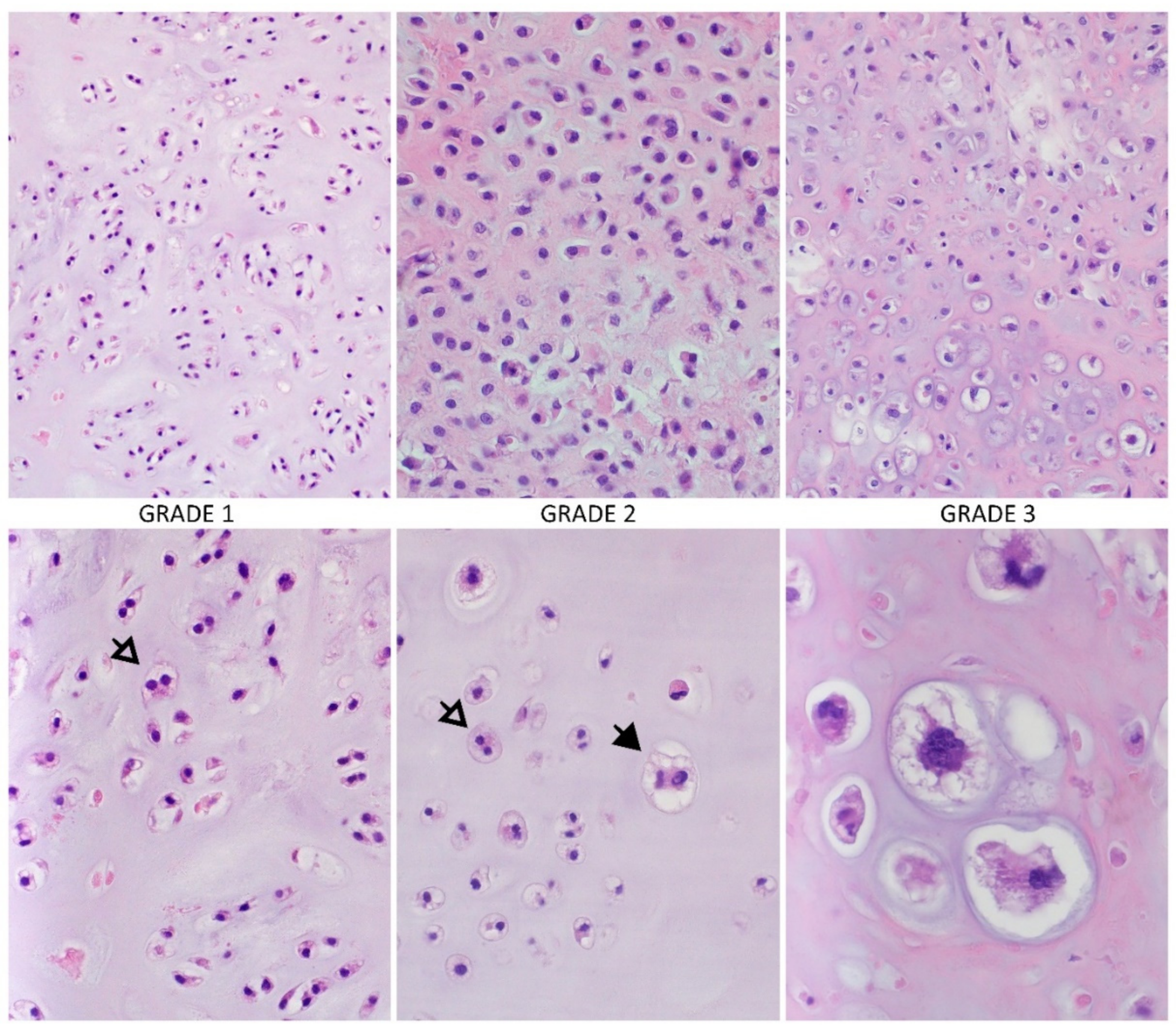
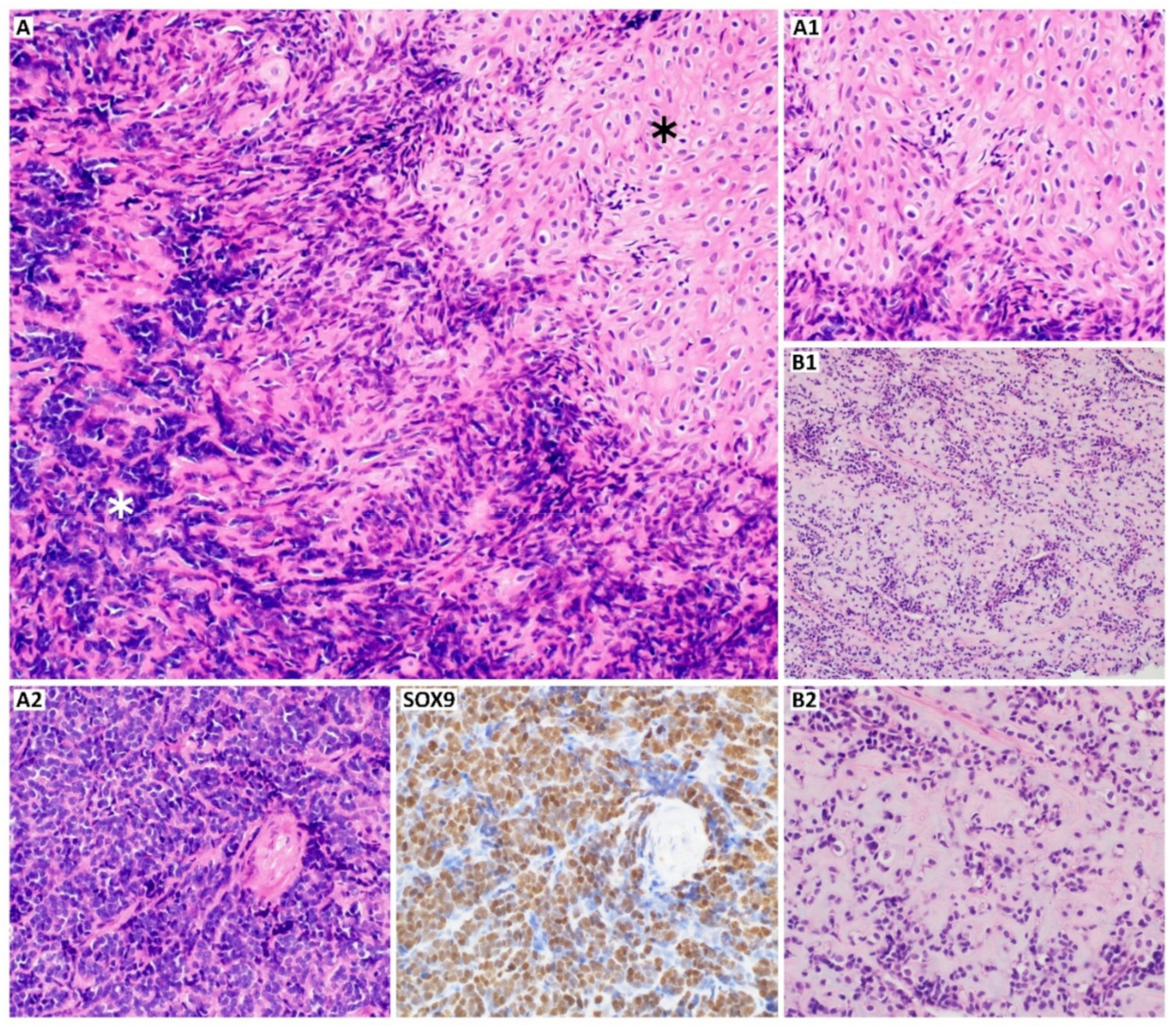
2. Morphology and Differentiation of Chondrosarcoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S100 | SOX9 | Bcl-2 | NY-ESO | P53 | Mutated IDH | Others | References | |
|---|---|---|---|---|---|---|---|---|
| Conventional | + | + | + | + | + (grade 2 and 3) | + | Brachyury, Col2a1, Cox-2, D2-40, Gal-1, MDM2, osteonectin, periostin, PTHrP, YKL-40 | [34,41,44,45,48,55,58,60,61,62,63,64] |
| Clear cell | + | + | + | − | + | − | Col2a1, keratine, Runx2 | [34,37,42,55,56] |
| Mesenchymal | + | + | + | − | + | − | CD99, desmin, EMA ezrin, MYF4, MYOD1, NKX2.2, vimentin | [30,31,34,55,56,57,60,63,65] |
| Dedifferentiated | − | + # | + | + | + * | + ** | CD44, Col1a1, Col2a1, cyclin D1, Ezrin, MDM2 *, PAI-1, PD-L1 ***, PTHR, Runx2 | [34,36,37,45,55,56,57,60,63,66] |
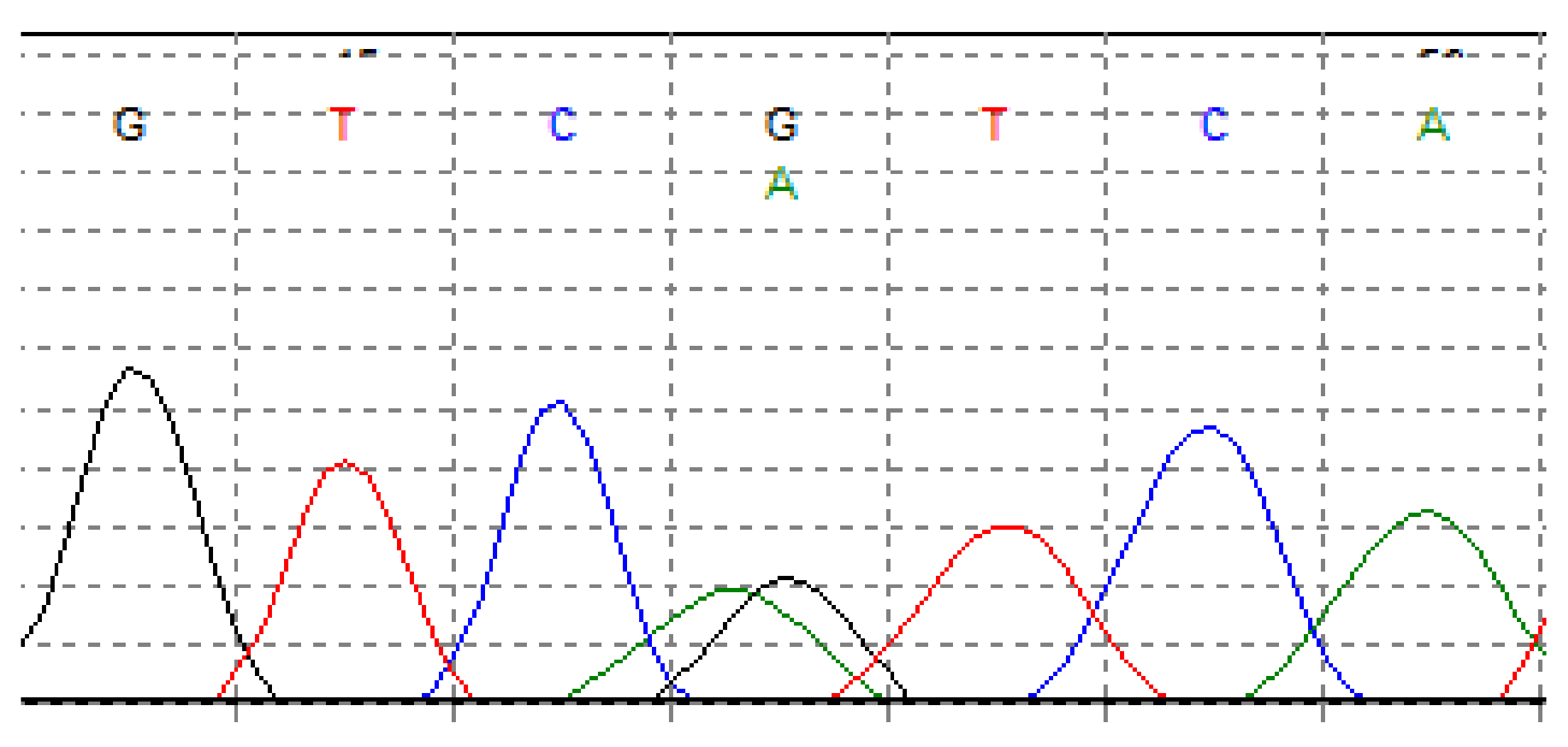
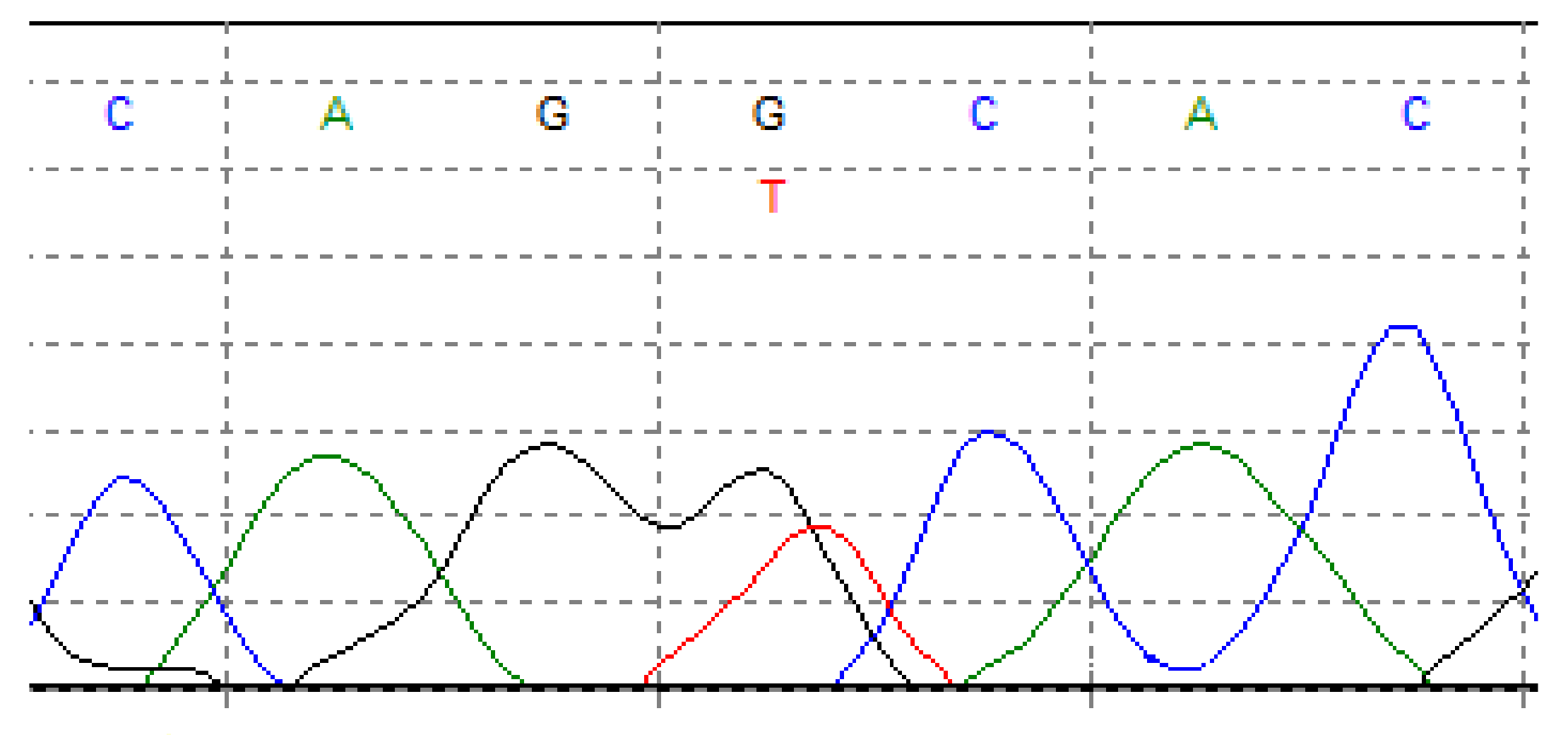
3. Genetics of Chondrosarcoma
4. Imaging Methods in Chondrosarcoma
5. Surgical Treatment of Chondrosarcoma
- Type I: limited to the ilium.
- Type II: limited to the periacetabulum.
- Type III: limited to the pubis.
6. Radiotherapy in Chondrosarcoma
7. Chondrosarcoma Chemotherapy
8. Targeted Therapies in Chondrosarcoma
8.1. Angiogenesis Inhibitors
8.2. Cyclin-Dependent Kinase Inhibitors
8.3. The Hedgehog Inhibitors
8.4. Histone Deacetylase Inhibitors
8.5. IDH Inhibitors
8.6. Tyrosine Kinase Inhibitors
8.7. mTOR Inhibitors
8.8. Osteoclast Inhibitors
8.9. Immunotherapy in Chondrosarcoma
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, P.P.; Moussallem, C.D.; Deavers, M.T. Secondary chondrosarcoma. J. Am. Acad. Orthop. Surg. 2010, 18, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Valery, P.C.; Laversanne, M.; Bray, F. Bone cancer incidence by morphological subtype: A global assessment. Cancer Causes Control. 2015, 26, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.S.; Alston, R.D.; Eden, T.O.; Geraci, M.; Birch, J.M. The contrasting age-incidence patterns of bone tumours in teenagers and young adults: Implications for aetiology. Int. J. Cancer 2012, 131, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef]
- Whelan, J.; McTiernan, A.; Cooper, N.; Wong, Y.K.; Francis, M.; Vernon, S.; Strauss, S.J. Incidence and survival of malignant bone sarcomas in England 1979–2007. Int. J. Cancer 2012, 131, E508–E517. [Google Scholar] [CrossRef]
- Anfinsen, K.P.; Devesa, S.S.; Bray, F.; Troisi, R.; Jonasdottir, T.J.; Bruland, O.S.; Grotmol, T. Age-period-cohort analysis of primary bone cancer incidence rates in the United States (1976–2005). Cancer Epidemiol. Biomark. Prev. 2011, 20, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Thorkildsen, J.; Taksdal, I.; Bjerkehagen, B.; Haugland, H.K.; Børge Johannesen, T.; Viset, T.; Norum, O.J.; Bruland, Ø.; Zaikova, O. Chondrosarcoma in Norway 1990–2013; an epidemiological and prognostic observational study of a complete national cohort. Acta Oncol. 2019, 58, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Van Praag Veroniek, V.M.; Rueten-Budde, A.J.; Ho, V.; Dijkstra, P.D.S.; Fiocco, M.; van de Sande, M.A.J. Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg. Oncol. 2018, 27, 402–408. [Google Scholar] [CrossRef]
- Polednak, A.P. Primary bone cancer incidence in black and white residents of New York State. Cancer 1985, 55, 2883–2888. [Google Scholar] [CrossRef]
- Dibas, M.; Doheim, M.F.; Ghozy, S.; Ros, M.H.; El-Helw, G.O.; Reda, A. Incidence and survival rates and trends of skull Base chondrosarcoma: A Population-Based study. Clin. Neurol. Neurosurg. 2020, 198, 106–153. [Google Scholar] [CrossRef]
- Amer, K.M.; Munn, M.; Congiusta, D.; Abraham, J.A.; Basu Mallick, A. Survival and Prognosis of Chondrosarcoma Subtypes: SEER Database Analysis. J. Orthop. Res. 2020, 38, 311–319. [Google Scholar] [CrossRef]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovee, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef]
- Tsuda, Y.; Gregory, J.J.; Fujiwara, T.; Abudu, S. Secondary chondrosarcoma arising from osteochondroma: Outcomes and prognostic factors. Bone Joint J. 2019, 101-b, 1313–1320. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Tan, T.S.; Unni, K.K.; Collins, M.S.; Wenger, D.E.; Sim, F.H. Secondary chondrosarcoma in osteochondroma: Report of 107 patients. Clin. Orthop. Relat. Res. 2003, 411, 193–206. [Google Scholar] [CrossRef]
- Nota, S.P.F.T.; Braun, Y.; Schwab, J.H.; van Dijk, C.N.; Bramer, J.A.M. The Identification of Prognostic Factors and Survival Statistics of Conventional Central Chondrosarcoma. Sarcoma 2015, 2015, 623746. [Google Scholar] [CrossRef] [Green Version]
- Skeletal Lesions Interobserver Correlation among Expert Diagnosticians (SLICED) Study Group. Reliability of histopathologic and radiologic grading of cartilaginous neoplasms in long bones. J. Bone Joint Surg. Am. 2007, 89, 2113–2123. [Google Scholar] [CrossRef]
- Thorkildsen, J.; Taksdal, I.; Bjerkehagen, B.; Norum, O.J.; Myklebust, T.A.; Zaikova, O. Risk stratification for central conventional chondrosarcoma of bone: A novel system predicting risk of metastasis and death in the Cancer Registry of Norway cohort. J. Surg. Oncol. 2020, 121, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Angelini, A.; Guerra, G.; Mavrogenis, A.F.; Pala, E.; Picci, P.; Ruggieri, P. Clinical outcome of central conventional chondrosarcoma. J. Surg. Oncol. 2012, 106, 929–937. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef]
- Bovée, J.V.M.G.; Bloem, J.L.; Flanagan, A.M.; Nielsen, G.P.; Yoshida, A. WHO Classification of Tumours: Soft Tissue and Bone Tumour, 5th ed.; WHO: Geneva, Switzerland, 2020; Volume 3, pp. 370–390. [Google Scholar]
- Nazeri, E.; Gouran Savadkoohi, M.; Majidzadeh-A, K.; Esmaeili, R. Chondrosarcoma: An overview of clinical behavior, molecular mechanisms mediated drug resistance and potential therapeutic targets. Crit. Rev. Oncol. Hematol. 2018, 131, 102–109. [Google Scholar] [CrossRef]
- Evans, H.L.; Ayala, A.G.; Romsdahl, M.M. Prognostic factors in chondrosarcoma of bone. A clinicopathologic analysis with emphasis on histologic grading. Cancer 1977, 40, 818–831. [Google Scholar] [CrossRef]
- Hogendoorn, P.B.; Bovee, J.M.; Bovee, J.M.; Nielsen, G.P. Chondrosarcoma (grades I-III), including primary and secondary variants and periosteal chondrosarcoma. In World Health Organization Classification of Tumours of Soft Tissue and Bone; Fletcher, C.D.M., Bridge, J.A., Hogendoorn, P.C.W., Mertens, F., Eds.; WHO: Geneva, Switzerland, 2013; Volume 5, p. 264. [Google Scholar]
- Fei, L.; Ngoh, C.; Porter, D.E. Chondrosarcoma transformation in hereditary multiple exostoses: A systematic review and clinical and cost-effectiveness of a proposed screening model. J. Bone Oncol. 2018, 13, 114–122. [Google Scholar] [CrossRef]
- Verdegaal, S.H.; Bovée, J.V.; Pansuriya, T.C.; Grimer, R.J.; Ozger, H.; Jutte, P.C.; San Julian, M.; Biau, D.J.; van der Geest, I.C.; Leithner, A.; et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicenter study of 161 patients. Oncologist 2011, 16, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- El Abiad, J.M.; Robbins, S.M.; Cohen, B.; Levin, A.S.; Valle, D.L.; Morris, C.D.; de Macena Sobreira, N.L. Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am. J. Med. Genet. A 2020, 182, 1093–1103. [Google Scholar] [CrossRef]
- Murphey, M.D.; Walker, E.A.; Wilson, A.J.; Kransdorf, M.J.; Temple, H.T.; Gannon, F.H. From the archives of the AFIP: Imaging of primary chondrosarcoma: Radiologic-pathologic correlation. Radiographics 2003, 23, 1245–1278. [Google Scholar] [CrossRef] [Green Version]
- Limaiem, F.; Davis, D.D.; Sticco, K.L. Chondrosarcoma; StatPearls Publishing LLC: Treasure Island, FL, USA, 2020. [Google Scholar]
- Chow, W.A. Chondrosarcoma: Biology, genetics, and epigenetics. F1000Res 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Syed, M.; Mushtaq, S.; Loya, A.; Hassan, U. NKX3.1 a useful marker for mesenchymal chondrosarcoma: An immunohistochemical study. Ann. Diagn. Pathol. 2020. [Google Scholar] [CrossRef]
- Fanburg-Smith, J.C.; Auerbach, A.; Marwaha, J.S.; Wang, Z.; Santi, M.; Judkins, A.R.; Rushing, E.J. Immunoprofile of mesenchymal chondrosarcoma: Aberrant desmin and EMA expression, retention of INI1, and negative estrogen receptor in 22 female-predominant central nervous system and musculoskeletal cases. Ann. Diagn. Pathol. 2010, 14, 8–14. [Google Scholar] [CrossRef]
- Fiedorowicz, M.; Bartnik, E.; Sobczuk, P.; Teterycz, P.; Czarnecka, A.M. Molecular biology of sarcoma. Oncol. Clin. Pract. 2018, 14, 307–330. [Google Scholar] [CrossRef]
- Dahlin, D.C.; Beabout, J.W. Dedifferentiation of low-grade chondrosarcomas. Cancer 1971, 28, 461–466. [Google Scholar] [CrossRef]
- Chebib, I.; Hornicek, F.J.; Bredella, M.A.; Deshpande, V.; Nielsen, G.P. Histologic variants of chondrosarcoma. Diagn. Histopath. 2014, 20, 172–180. [Google Scholar] [CrossRef]
- Kashima, T.G.; Dongre, A.; Oppermann, U.; Athanasou, N.A. Dentine matrix protein 1 (DMP-1) is a marker of bone-forming tumours. Virchows Arch. 2013, 462, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.; Italiano, A.; Cleton-Jansen, A.M.; Bovée, J.V. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, D.; de Jong, D.; Pansuriya, T.C.; van den Akker, B.E.; Picci, P.; Szuhai, K.; Bovée, J.V.G.M. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes, Chromosomes Cancer 2012, 51, 899–909. [Google Scholar] [CrossRef]
- Mocellin, S. Extraskeletal Myxoid Chondrosarcoma. In Soft Tissue Tumors; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Rozeman, L.B.; Cleton-Jansen, A.M.; Hogendoorn, P.C.W. Pathology of primary malignant bone and cartilage tumours. Int. Orthop. 2006, 30, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Aigner, T.; Dertinger, S.; Belke, J.; Kirchner, T. Chondrocytic cell differentiation in clear cell chondrosarcoma. Hum. Pathol. 1996, 27, 1301–1305. [Google Scholar] [CrossRef]
- Cai, L.; Gao, Z.F.; Huang, X.Y. Clinicopathology analysis of mesenchymal chondrosarcoma in soft tissue. Beijing Da Xue Xue Bao Yi Xue Ban 2006, 38, 501–505. [Google Scholar]
- Jeong, W.; Kim, H.-J. Biomarkers of chondrosarcoma. J. Clin. Pathol 2018, 71, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lu, X.; Guo, W.; Ren, T.; Zhao, H.; Zhao, F.; Tang, G. Different expression of Sox9 and Runx2 between chondrosarcoma and dedifferentiated chondrosarcoma cell line. Eur. J. Cancer Prev. 2010, 19, 466–471. [Google Scholar] [CrossRef]
- Shen, J.; Meyers, C.A.; Shrestha, S.; Singh, A.; LaChaud, G.; Nguyen, V.; Asatrian, G.; Federman, N.; Bernthal, N.; Eilber, F.C.; et al. Sclerostin expression in skeletal sarcomas. Hum. Pathol. 2016, 58, 24–34. [Google Scholar] [CrossRef]
- Lai, X.; Chen, S. Identification of novel biomarker candidates for immunohistochemical diagnosis to distinguish low-grade chondrosarcoma from enchondroma. Proteomics 2015, 15, 2358–2368. [Google Scholar] [CrossRef] [Green Version]
- Boehme, K.A.; Schleicher, S.B.; Traub, F.; Rolauffs, B. Chondrosarcoma: A Rare Misfortune in Aging Human Cartilage? The Role of Stem and Progenitor Cells in Proliferation, Malignant Degeneration and Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 311. [Google Scholar] [CrossRef] [Green Version]
- Wehrli, B.M.; Huang, W.; De Crombrugghe, B.; Ayala, A.G.; Czerniak, B. Sox9, a master regulator of chondrogenesis, distinguishes mesenchymal chondrosarcoma from other small blue round cell tumors. Hum. Pathol. 2003, 34, 263–269. [Google Scholar] [CrossRef]
- Cajaiba, M.M.; Jianhua, L.; Goodman, M.A.; Fuhrer, K.A.; Rao, U.N. Sox9 expression is not limited to chondroid neoplasms: Variable occurrence in other soft tissue and bone tumors with frequent expression by synovial sarcomas. Int. J. Surg. Pathol. 2010, 18, 319–323. [Google Scholar] [CrossRef]
- Konishi, E.; Nakashima, Y.; Iwasa, Y.; Nakao, R.; Yanagisawa, A. Immunohistochemical analysis for Sox9 reveals the cartilaginous character of chondroblastoma and chondromyxoid fibroma of the bone. Hum. Pathol. 2010, 41, 208–213. [Google Scholar] [CrossRef]
- Zhu, H.; Tang, J.; Tang, M.; Cai, H. Upregulation of SOX9 in osteosarcoma and its association with tumor progression and patients’ prognosis. Diagn. Pathol. 2013, 8, 183. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.F.; Hayes, M.M.; Lebrun, D.; Espinosa, I.; Nielsen, G.P.; Rosenberg, A.E.; Lee, C.H. FLI-1 distinguishes Ewing sarcoma from small cell osteosarcoma and mesenchymal chondrosarcoma. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 233–238. [Google Scholar] [CrossRef]
- Machado, I.; Alberghini, M.; Giner, F.; Corrigan, M.; O’Sullivan, M.; Noguera, R.; Pellin, A.; Bertoni, F.; Llombart-Bosch, A. Histopathological characterization of small cell osteosarcoma with immunohistochemistry and molecular genetic support. A study of 10 cases. Histopathology 2010, 57, 162–167. [Google Scholar] [CrossRef]
- Endo, M.; de Graaff, M.A.; Ingram, D.R.; Lim, S.; Lev, D.C.; Briaire-de Bruijn, I.H.; Somaiah, N.; Bovée, J.V.M.G.; Lazar, A.J.; Nielsen, T.O. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Modern Pathol. 2015, 28, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Van Oosterwijk, J.G.; Meijer, D.; van Ruler, M.A.; van den Akker, B.E.; Oosting, J.; Krenacs, T.; Picci, P.; Flanagan, A.M.; Liegl-Atzwanger, B.; Leithner, A.; et al. Screening for potential targets for therapy in mesenchymal, clear cell, and dedifferentiated chondrosarcoma reveals Bcl-2 family members and TGFbeta as potential targets. Am. J. Pathol. 2013, 182, 1347–1356. [Google Scholar] [CrossRef]
- Salas, S.; de Pinieux, G.; Gomez-Brouchet, A.; Larrousserie, F.; Leroy, X.; Aubert, S.; Decouvelaere, A.V.; Giorgi, R.; Fernandez, C.; Bouvier, C. Ezrin immunohistochemical expression in cartilaginous tumours: A useful tool for differential diagnosis between chondroblastic osteosarcoma and chondrosarcoma. Virchows Arch. 2009, 454, 81–87. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Mourcin, F.; Gourraud, P.A.; Bouvier, C.; De Pinieux, G.; Le Guelec, S.; Brousset, P.; Delisle, M.B.; Schiff, C. Galectin-1 is a powerful marker to distinguish chondroblastic osteosarcoma and conventional chondrosarcoma. Hum. Pathol. 2010, 41, 1220–1230. [Google Scholar] [CrossRef]
- Taddei, M.L.; Pietrovito, L.; Leo, A.; Chiarugi, P. Lactate in Sarcoma Microenvironment: Much More than just a Waste Product. Cells 2020, 9, 510. [Google Scholar] [CrossRef] [Green Version]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Daugaard, S.; Christensen, L.H.; Høgdall, E. Markers aiding the diagnosis of chondroid tumors: An immunohistochemical study including osteonectin, bcl-2, cox-2, actin, calponin, D2-40 (podoplanin), mdm-2, CD117 (c-kit), and YKL-40. APMIS 2009, 117, 518–525. [Google Scholar] [CrossRef] [Green Version]
- De Jong, Y.; van Oosterwijk, J.G.; Kruisselbrink, A.B.; Briaire-de Bruijn, I.H.; Agrogiannis, G.; Baranski, Z.; Cleven, A.H.G.; Cleton-Jansen, A.-M.; van de Water, B.; Danen, E.H.J.; et al. Targeting survivin as a potential new treatment for chondrosarcoma of bone. Oncogenesis 2016, 5. [Google Scholar] [CrossRef]
- Kim, M.-J.; Cho, K.-J.; Ayala, A.G.; Ro, J.Y. Chondrosarcoma: With updates on molecular genetics. Sarcoma 2011, 2011, 405437. [Google Scholar] [CrossRef] [Green Version]
- Oakley, G.J.; Fuhrer, K.; Seethala, R.R. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: A tissue microarray-based comparative analysis. Modern Pathol. 2008, 21, 1461–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folpe, A.L.; Graham, R.P.; Martinez, A.; Schembri-Wismayer, D.; Boland, J.; Fritchie, K.J. Mesenchymal chondrosarcomas showing immunohistochemical evidence of rhabdomyoblastic differentiation: A potential diagnostic pitfall. Hum. Pathol. 2018, 77, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Stöckl, S.; Lindner, G.; Li, S.; Schuster, P.; Haferkamp, S.; Wagner, F.; Prodinger, P.M.; Multhoff, G.; Boxberg, M.; Hillmann, A.; et al. SOX9 Knockout Induces Polyploidy and Changes Sensitivity to Tumor Treatment Strategies in a Chondrosarcoma Cell Line. Int. J. Mol. Sci. 2020, 21, 7627. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef]
- Tallegas, M.; Miquelestorena-Standley, É.; Labit-Bouvier, C.; Badoual, C.; Francois, A.; Gomez-Brouchet, A.; Aubert, S.; Collin, C.; Tallet, A.; de Pinieux, G. IDH mutation status in a series of 88 head and neck chondrosarcomas: Different profile between tumors of the skull base and tumors involving the facial skeleton and the laryngotracheal tract. Hum. Pathol. 2019, 84, 183–191. [Google Scholar] [CrossRef]
- Tap, W.D.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.J.; Jiang, L.; Wu, B.; et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients With Advanced Chondrosarcoma. J. Clin. Oncol. 2020, 38, 1693–1701. [Google Scholar] [CrossRef]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hu, X.; Eid, J.E.; Rosenberg, A.E.; Wilky, B.A.; Ban, Y.; Sun, X.; Galoian, K.; DeSalvo, J.; Yue, J.; et al. Mutant IDH1 Depletion Downregulates Integrins and Impairs Chondrosarcoma Growth. Cancers 2020, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Fritchie, K.; Wei, S.; Ali, N.; Curless, K.; Shen, T.; Brini, A.T.; Latif, F.; Sumathi, V.; Siegal, G.P.; et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum. Pathol. 2017, 65, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Bovée, J.V.; Hogendoorn, P.C.; Wunder, J.S.; Alman, B.A. Cartilage tumours and bone development: Molecular pathology and possible therapeutic targets. Nat. Rev. Cancer 2010, 10, 481–488. [Google Scholar] [CrossRef]
- Asp, J.; Inerot, S.; Block, J.A.; Lindahl, A. Alterations in the regulatory pathway involving p16, pRb and cdk4 in human chondrosarcoma. J. Orthop. Res. 2001, 19, 149–154. [Google Scholar] [CrossRef]
- Dobashi, Y.; Sugimura, H.; Sato, A.; Hirabayashi, T.; Kanda, H.; Kitagawa, T.; Kawaguchi, N.; Imamura, T.; Machinami, R. Possible association of p53 overexpression and mutation with high-grade chondrosarcoma. Diagn. Mol. Pathol. 1993, 2, 257–263. [Google Scholar] [CrossRef]
- Schrage, Y.M.; Lam, S.; Jochemsen, A.G.; Cleton-Jansen, A.-M.; Taminiau, A.H.M.; Hogendoorn, P.C.W.; Bovée, J.V.M.G. Central chondrosarcoma progression is associated with pRb pathway alterations: CDK4 down-regulation and p16 overexpression inhibit cell growth in vitro. J. Cell Mol. Med. 2009, 13, 2843–2852. [Google Scholar] [CrossRef]
- Cleven, A.H.; Zwartkruis, E.; Hogendoorn, P.C.; Kroon, H.M.; Briaire-de Bruijn, I.; Bovee, J.V. Periosteal chondrosarcoma: A histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology 2015, 67, 483–490. [Google Scholar] [CrossRef]
- Amary, M.F.; Ye, H.; Forbes, G.; Damato, S.; Maggiani, F.; Pollock, R.; Tirabosco, R.; Flanagan, A.M. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015, 466, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.; Radmacher, M.; Mohammed, N.; Suster, D.; Auer, H.; Jones, S.; Riggenbach, J.; Kelbick, N.; Bos, G.; Mayerson, J. MYC amplification and polysomy 8 in chondrosarcoma: Array comparative genomic hybridization, fluorescent in situ hybridization, and association with outcome. J. Clin. Oncol. 2005, 23, 9369–9376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallor, K.H.; Staaf, J.; Bovee, J.V.M.G.; Hogendoorn, P.C.W.; Cleton-Jansen, A.-M.; Knuutila, S.; Savola, S.; Niini, T.; Brosjo, O.; Bauer, H.C.F.; et al. Genomic Profiling of Chondrosarcoma: Chromosomal Patterns in Central and Peripheral Tumors. Clinical Cancer Res. 2009, 15, 2685–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, J.H.; Boland, P.J.; Agaram, N.P.; Socci, N.D.; Guo, T.; O’Toole, G.C.; Wang, X.; Ostroumov, E.; Hunter, C.J.; Block, J.A.; et al. Chordoma and chondrosarcoma gene profile: Implications for immunotherapy. Cancer Immunol. Immunother. 2008, 58, 339. [Google Scholar] [CrossRef] [Green Version]
- Bovee, J.V.; Cleton-Jansen, A.M.; Kuipers-Dijkshoorn, N.J.; van den Broek, L.J.; Taminiau, A.H.; Cornelisse, C.J.; Hogendoorn, P.C. Loss of heterozygosity and DNA ploidy point to a diverging genetic mechanism in the origin of peripheral and central chondrosarcoma. Genes Chromosomes Cancer 1999, 26, 237–246. [Google Scholar] [CrossRef]
- De Andrea, C.E.; Reijnders, C.M.; Kroon, H.M.; de Jong, D.; Hogendoorn, P.C.; Szuhai, K.; Bovee, J.V. Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene 2012, 31, 1095–1104. [Google Scholar] [CrossRef]
- Cote, G.M.; He, J.; Choy, E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist 2018, 23, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shon, W.; Folpe, A.L.; Fritchie, K.J. ERG expression in chondrogenic bone and soft tissue tumours. J. Clin. Pathol 2015, 68, 125–129. [Google Scholar] [CrossRef]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Boye, K.; Heim, S. Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncol. Rep. 2014, 32, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Haugom, L.; Gorunova, L.; Bjerkehagen, B.; Fosså, A.; Guriby, M.; Nome, T.; Lothe, R.A.; et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS ONE 2012, 7, e49705. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Baldi, G.G.; Morosi, C.; Gronchi, A.; Maestro, R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers 2020, 12, 2703. [Google Scholar] [CrossRef]
- Leddy, L.R.; Holmes, R.E. Chondrosarcoma of bone. Cancer Treat. Res. 2014, 162, 117–130. [Google Scholar] [CrossRef]
- Giuffrida, A.Y.; Burgueno, J.E.; Koniaris, L.G.; Gutierrez, J.C.; Duncan, R.; Scully, S.P. Chondrosarcoma in the United States (1973 to 2003): An analysis of 2890 cases from the SEER database. J. Bone Joint Surg. Am. 2009, 91, 1063–1072. [Google Scholar] [CrossRef]
- Stevenson, J.D.; Laitinen, M.K.; Parry, M.C.; Sumathi, V.; Grimer, R.J.; Jeys, L.M. The role of surgical margins in chondrosarcoma. Eur. J. Surg. Oncol. 2018, 44, 1412–1418. [Google Scholar] [CrossRef] [Green Version]
- De Souza, L.L.; Pontes, F.S.C.; Fonseca, F.P.; da Mata Rezende, D.S.; Vasconcelos, V.C.S.; Pontes, H.A.R. Chondrosarcoma of the jaw bones: A review of 224 cases reported to date and an analysis of prognostic factors. Int. J. Oral Maxillofac. Surg. 2019, 48, 452–460. [Google Scholar] [CrossRef]
- Arora, K.; Riddle, N.D. Extraskeletal Mesenchymal Chondrosarcoma. Arch. Pathol. Lab. Med. 2018, 142, 1421–1424. [Google Scholar] [CrossRef]
- Chin, O.Y.; Dubal, P.M.; Sheikh, A.B.; Unsal, A.A.; Park, R.C.; Baredes, S.; Eloy, J.A. Laryngeal chondrosarcoma: A systematic review of 592 cases. Laryngoscope 2017, 127, 430–439. [Google Scholar] [CrossRef]
- Soldatos, T.; McCarthy, E.F.; Attar, S.; Carrino, J.A.; Fayad, L.M. Imaging features of chondrosarcoma. J. Comput. Assist. Tomogr. 2011, 35, 504–511. [Google Scholar] [CrossRef]
- Ollivier, L.; Vanel, D.; Leclere, J. Imaging of chondrosarcomas. Cancer Imaging 2003, 4, 36–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roitman, P.D.; Farfalli, G.L.; Ayerza, M.A.; Múscolo, D.L.; Milano, F.E.; Aponte-Tinao, L.A. Is Needle Biopsy Clinically Useful in Preoperative Grading of Central Chondrosarcoma of the Pelvis and Long Bones? Clin. Orthop. Relat. Res. 2017, 475, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Herget, G.W.; Uhl, M.; Opitz, O.G.; Adler, C.P.; Südkamp, N.P.; Knöller, S. The many faces of chondrosarcoma of bone, own cases and review of the literature with an emphasis on radiology, pathology and treatment. Acta Chir. Orthop. Traumatol. Cech. 2011, 78, 501–509. [Google Scholar] [PubMed]
- Littrell, L.A.; Wenger, D.E.; Wold, L.E.; Bertoni, F.; Unni, K.K.; White, L.M.; Kandel, R.; Sundaram, M. Radiographic, CT, and MR imaging features of dedifferentiated chondrosarcomas: A retrospective review of 174 de novo cases. Radiographics 2004, 24, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphey, M.D.; Flemming, D.J.; Boyea, S.R.; Bojescul, J.A.; Sweet, D.E.; Temple, H.T. Enchondroma versus chondrosarcoma in the appendicular skeleton: Differentiating features. Radiographics 1998, 18, 1213–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhumaid, S.M.; Alharbi, A.T.; Aljubair, H. Magnetic Resonance Imaging Role in the Differentiation Between Atypical Cartilaginous Tumors and High-Grade Chondrosarcoma: An Updated Systematic Review. Cureus 2020, 12, e11237. [Google Scholar] [CrossRef]
- Fabbri, N.; Donati, D. Chondrosarcomas (CHS). In Atlas of Musculoskeletal Tumors and Tumorlike Lesions: The Rizzoli Case Archive; Picci, P., Manfrini, M., Fabbri, N., Gambarotti, M., Vanel, D., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 111–112. [Google Scholar] [CrossRef]
- De Coninck, T.; Jans, L.; Sys, G.; Huysse, W.; Verstraeten, T.; Forsyth, R.; Poffyn, B.; Verstraete, K. Dynamic contrast-enhanced MR imaging for differentiation between enchondroma and chondrosarcoma. Eur. Radiol. 2013, 23, 3140–3152. [Google Scholar] [CrossRef]
- Douis, H.; Parry, M.; Vaiyapuri, S.; Davies, A.M. What are the differentiating clinical and MRI-features of enchondromas from low-grade chondrosarcomas? Eur. Radiol. 2018, 28, 398–409. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, K.; Ding, Y.; Liang, W.; Ding, Y.; Vanel, D.; Cheng, X. Study of single voxel 1H MR spectroscopy of bone tumors: Differentiation of benign from malignant tumors. Eur. Radiol. 2013, 82, 2124–2128. [Google Scholar] [CrossRef]
- Fayad, L.M.; Bluemke, D.A.; McCarthy, E.F.; Weber, K.L.; Barker, P.B.; Jacobs, M.A. Musculoskeletal tumors: Use of proton MR spectroscopic imaging for characterization. JMRI 2006, 23, 23–28. [Google Scholar] [CrossRef]
- Yao, K.; Troupis, J.M. Diffusion-weighted imaging and the skeletal system: A literature review. Clin. Radiol. 2016, 71, 1071–1082. [Google Scholar] [CrossRef]
- Müller, U.; Kubik-Huch, R.A.; Ares, C.; Hug, E.B.; Löw, R.; Valavanis, A.; Ahlhelm, F.J. Is there a role for conventional MRI and MR diffusion-weighted imaging for distinction of skull base chordoma and chondrosarcoma? Acta Radiol. 2016, 57, 225–232. [Google Scholar] [CrossRef]
- Hayashida, Y.; Hirai, T.; Yakushiji, T.; Katahira, K.; Shimomura, O.; Imuta, M.; Nakaura, T.; Utsunomiya, D.; Awai, K.; Yamashita, Y. Evaluation of diffusion-weighted imaging for the differential diagnosis of poorly contrast-enhanced and T2-prolonged bone masses: Initial experience. J. Magn. Reson. Imaging 2006, 23, 377–382. [Google Scholar] [CrossRef]
- Yin, P.; Mao, N.; Liu, X.; Sun, C.; Wang, S.; Chen, L.; Hong, N. Can clinical radiomics nomogram based on 3D multiparametric MRI features and clinical characteristics estimate early recurrence of pelvic chondrosarcoma? J. Magn. Reson. Imaging 2020, 51, 435–445. [Google Scholar] [CrossRef]
- Jo, I.; Gould, D.; Schlicht, S.; Taubman, K.; Choong, P. Diagnostic accuracy of functional imaging modalities for chondrosarcoma: A systematic review and meta-analysis. J. Bone Oncol. 2019, 19, 100262. [Google Scholar] [CrossRef]
- Choi, W.H.; Han, E.J.; Chang, K.B.; Joo, M.W. Quantitative SPECT/CT for differentiating between enchondroma and grade I chondrosarcoma. Sci. Rep. 2020, 10, 10587. [Google Scholar] [CrossRef]
- Jo, O.; Schlicht, S.; Slavin, J.; Di Bella, C.; Pang, G.; Powell, G.; Spelman, T.; Choong, P.F. The role of Thallium-201 scintigraphy and Tc-99m pentavalent dimercaptosuccinic acid in diagnosis and grading of chondrosarcoma. Eur.J. Radiol. 2020, 125, 108846. [Google Scholar] [CrossRef]
- Subhawong, T.K.; Winn, A.; Shemesh, S.S.; Pretell-Mazzini, J. F-18 FDG PET differentiation of benign from malignant chondroid neoplasms: A systematic review of the literature. Skeletal Radiol. 2017, 46, 1233–1239. [Google Scholar] [CrossRef]
- Zhang, Q.; Xi, Y.; Li, D.; Yuan, Z.; Dong, J. The utility of (18)F-FDG PET and PET/CT in the diagnosis and staging of chondrosarcoma: A meta-analysis. J. Orthop. Surg. Res. 2020, 15, 229. [Google Scholar] [CrossRef]
- Laitinen, M.K.; Parry, M.C.; Le Nail, L.R.; Wigley, C.H.; Stevenson, J.D.; Jeys, L.M. Locally recurrent chondrosarcoma of the pelvis and limbs can only be controlled by wide local excision. Bone Joint J. 2019, 101-B, 266–271. [Google Scholar] [CrossRef]
- Wirbel, R.J.; Schulte, M.; Maier, B.; Koschnik, M.; Mutschler, W.E. Chondrosarcoma of the pelvis: Oncologic and functional outcome. Sarcoma 2000, 4, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Jamil, N.; Howie, S.; Salter, D.M. Therapeutic molecular targets in human chondrosarcoma. Int. J. Exp. Pathol. 2010, 91, 387–393. [Google Scholar] [CrossRef]
- Chen, X.; Yu, L.J.; Peng, H.M.; Jiang, C.; Ye, C.H.; Zhu, S.B.; Qian, W.W. Is intralesional resection suitable for central grade 1 chondrosarcoma: A systematic review and updated meta-analysis. Eur. J. Surg. Oncol. 2017, 43, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Fiorenza, F.; Abudu, A.; Grimer, R.J.; Carter, S.R.; Tillman, R.M.; Ayoub, K.; Mangham, D.C.; Davies, A.M. Risk factors for survival and local control in chondrosarcoma of bone. J. Bone Joint Surg. Br. 2002, 84, 93–99. [Google Scholar] [CrossRef]
- Donati, D.; El Ghoneimy, A.; Bertoni, F.; Di Bella, C.; Mercuri, M. Surgical treatment and outcome of conventional pelvic chondrosarcoma. J. Bone Joint Surg. Br. 2005, 87, 1527–1530. [Google Scholar] [CrossRef]
- Enneking, W.F.; Dunham, W.K. Resection and reconstruction for primary neoplasms involving the innominate bone. J. Bone Joint Surg. Am. 1978, 60, 731–746. [Google Scholar] [CrossRef]
- Fujiwara, T.; Kaneuchi, Y.; Stevenson, J.; Parry, M.; Kurisunkal, V.; Clark, R.; Tsuda, Y.; Laitinen, M.; Grimer, R.; Jeys, L. Navigation-assisted pelvic resections and reconstructions for periacetabular chondrosarcomas. Eur. J. Surg. Oncol. 2021, 47, 416–423. [Google Scholar] [CrossRef]
- Jeys, L.; Matharu, G.S.; Nandra, R.S.; Grimer, R.J. Can computer navigation-assisted surgery reduce the risk of an intralesional margin and reduce the rate of local recurrence in patients with a tumour of the pelvis or sacrum? Bone Joint J. 2013, 95-B, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, M.K.; Parry, M.C.; Albergo, J.I.; Grimer, R.J.; Jeys, L.M. Is computer navigation when used in the surgery of iliosacral pelvic bone tumours safer for the patient? Bone Joint J. 2017, 99-B, 261–266. [Google Scholar] [CrossRef]
- Krettek, C.; Geerling, J.; Bastian, L.; Citak, M.; Rucker, F.; Kendoff, D.; Hufner, T. Computer aided tumor resection in the pelvis. Injury 2004, 35, A79–A83. [Google Scholar] [CrossRef]
- Leerapun, T.; Hugate, R.R.; Inwards, C.Y.; Scully, S.P.; Sim, F.H. Surgical management of conventional grade I chondrosarcoma of long bones. Clin. Orthop. Relat. Res. 2007, 463, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R., Jr.; Lautenschlager, E.P.; Moore, B.K. On the setting properties of acrylic bone cement. J. Bone Joint Surg. Am. 1973, 55, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Bingold, A.C.; Percy, A.J. Prosthetic replacement of a chondrosarcoma of the upper end of the femur: A 43-year follow-up. J. Bone Joint Surg. Br. 1996, 78, 663–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingold, A.C. Prosthetic replacement of a chondrosarcoma of the upper end of the femur. Eighteen-year follow-up. J. Bone Joint Surg. Br. 1972, 54, 139–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Bindiganavile, S.S.; Han, I. Oncologic outcome after local recurrence of chondrosarcoma: Analysis of prognostic factors. J. Surg. Oncol. 2015, 111, 957–961. [Google Scholar] [CrossRef] [PubMed]
- De Jong, Y.; Ingola, M.; Briaire-de Bruijn, I.H.; Kruisselbrink, A.B.; Venneker, S.; Palubeckaite, I.; Heijs, B.P.A.M.; Cleton-Jansen, A.-M.; Haas, R.L.M.; Bovée, J.V.M.G. Radiotherapy resistance in chondrosarcoma cells; a possible correlation with alterations in cell cycle related genes. Clin. Sarcoma Res. 2019, 9, 9. [Google Scholar] [CrossRef]
- Lechler, P.; Renkawitz, T.; Campean, V.; Balakrishnan, S.; Tingart, M.; Grifka, J.; Schaumburger, J. The antiapoptotic gene survivin is highly expressed in human chondrosarcoma and promotes drug resistance in chondrosarcoma cells in vitro. BMC Cancer 2011, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network: Bone cancer (2019). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1418 (accessed on 11 April 2021).
- Scandinavian Sarcoma Group. SSG XXIV Recommendations for Radiotherapy in Bone- and Soft Tissue Sarcoma. Available online: http://www.ssg-org.net/wp-content/uploads/2011/05/SSG-RT-Guidelines-December-2015.pdf (accessed on 11 April 2021).
- Redondo, A.; Bagué, S.; Bernabeu, D.; Ortiz-Cruz, E.; Valverde, C.; Alvarez, R.; Martinez-Trufero, J.; Lopez-Martin, J.A.; Correa, R.; Cruz, J.; et al. Malignant bone tumors (other than Ewing’s): Clinical practice guidelines for diagnosis, treatment and follow-up by Spanish Group for Research on Sarcomas (GEIS). Cancer Chemother. Pharmacol. 2017, 80, 1113–1131. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.G.; Jian, B.J.; Yang, I.; Han, S.J.; Aranda, D.; Ahn, B.J.; Parsa, A.T. Cranial chondrosarcoma and recurrence. Skull Base 2010, 20, 149–156. [Google Scholar] [CrossRef] [Green Version]
- DeLaney, T.F.; Liebsch, N.J.; Pedlow, F.X.; Adams, J.; Weyman, E.A.; Yeap, B.Y.; Depauw, N.; Nielsen, G.P.; Harmon, D.C.; Yoon, S.S.; et al. Long-term results of Phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J. Surg. Oncol. 2014, 110, 115–122. [Google Scholar] [CrossRef]
- Potluri, S.; Jefferies, S.J.; Jena, R.; Harris, F.; Burton, K.E.; Prevost, A.T.; Burnet, N.G. Residual postoperative tumour volume predicts outcome after high-dose radiotherapy for chordoma and chondrosarcoma of the skull base and spine. Clin. Oncol. 2011, 23, 199–208. [Google Scholar] [CrossRef]
- Oike, T.; Niimi, A.; Okonogi, N.; Murata, K.; Matsumura, A.; Noda, S.-E.; Kobayashi, D.; Iwanaga, M.; Tsuchida, K.; Kanai, T.; et al. Visualization of complex DNA double-strand breaks in a tumor treated with carbon ion radiotherapy. Sci. Rep. 2016, 6, 22275. [Google Scholar] [CrossRef] [Green Version]
- Ares, C.; Hug, E.B.; Lomax, A.J.; Bolsi, A.; Timmermann, B.; Rutz, H.P.; Schuller, J.C.; Pedroni, E.; Goitein, G. Effectiveness and safety of spot scanning proton radiation therapy for chordomas and chondrosarcomas of the skull base: First long-term report. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 1111–1118. [Google Scholar] [CrossRef]
- Bhatt, A.D.; Jacobson, A.; Lee, R.Y.; Giraud, C.; Schwab, J.H.; Hornicek, F.J.; Nielsen, P.; Choy, E.; Harmon, D.; DeLaney, T.F.; et al. High-Dose Proton Beam–Based Radiation Therapy in the Management of Extracranial Chondrosarcomas. Int. J. Part. Ther. 2016, 3, 373–381. [Google Scholar] [CrossRef]
- Guan, X.; Gao, J.; Hu, J.; Hu, W.; Yang, J.; Qiu, X.; Hu, C.; Kong, L.; Lu, J.J. The preliminary results of proton and carbon ion therapy for chordoma and chondrosarcoma of the skull base and cervical spine. Radiat. Oncol. 2019, 14, 206. [Google Scholar] [CrossRef]
- Habrand, J.L.; Schneider, R.; Alapetite, C.; Feuvret, L.; Petras, S.; Datchary, J.; Grill, J.; Noel, G.; Helfre, S.; Ferrand, R.; et al. Proton therapy in pediatric skull base and cervical canal low-grade bone malignancies. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 672–675. [Google Scholar] [CrossRef]
- Holtzman, A.L.; Rotondo, R.L.; Rutenberg, M.S.; Indelicato, D.J.; Mercado, C.E.; Rao, D.; Tavanaiepour, D.; Morris, C.G.; Louis, D.; Flampouri, S.; et al. Proton therapy for skull-base chondrosarcoma, a single-institution outcomes study. J. Neurooncol. 2019, 142, 557–563. [Google Scholar] [CrossRef]
- Noel, G.; Habrand, J.L.; Jauffret, E.; de Crevoisier, R.; Dederke, S.; Mammar, H.; Haie-Meder, C.; Pontvert, D.; Hasboun, D.; Ferrand, R.; et al. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther. Onkol. 2003, 179, 241–248. [Google Scholar] [CrossRef]
- Palm, R.F.; Oliver, D.E.; Yang, G.Q.; Abuodeh, Y.; Naghavi, A.O.; Johnstone, P.A.S. The role of dose escalation and proton therapy in perioperative or definitive treatment of chondrosarcoma and chordoma: An analysis of the National Cancer Data Base. Cancer 2019, 125, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Ertner, D.; Nikoghosyan, A.; Hof, H.; Didinger, B.; Combs, S.E.; Jakel, O.; Karger, C.P.; Edler, L.; Debus, J. Carbon ion radiotherapy of skull base chondrosarcomas. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 171–177. [Google Scholar] [CrossRef]
- Weber, D.C.; Rutz, H.P.; Pedroni, E.S.; Bolsi, A.; Timmermann, B.; Verwey, J.; Lomax, A.J.; Goitein, G. Results of spot-scanning proton radiation therapy for chordoma and chondrosarcoma of the skull base: The Paul Scherrer Institut experience. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 401–409. [Google Scholar] [CrossRef]
- Andrä, C.; Eze, C.; Ganswindt, U.; Roeder, F.; Belka, C.; Ostermann, H.; Manapov, F. Simultaneous bilateral stereotactic body radiation therapy of two inoperable centrally located pulmonary lesions in a patient with metastatic mesenchymal chondrosarcoma. Cancer Treat. Commun. 2016, 6, 8–10. [Google Scholar] [CrossRef]
- Lindsay, A.D.; Haupt, E.E.; Chan, C.M.; Spiguel, A.R.; Scarborough, M.T.; Zlotecki, R.A.; Gibbs, P.C. Treatment of Sarcoma Lung Metastases with Stereotactic Body Radiotherapy. Sarcoma 2018, 2018, 9132359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasudevan, H.N.; Raleigh, D.R.; Johnson, J.; Garsa, A.A.; Theodosopoulos, P.V.; Aghi, M.K.; Ames, C.; McDermott, M.W.; Barani, I.J.; Braunstein, S.E. Management of Chordoma and Chondrosarcoma with Fractionated Stereotactic Radiotherapy. Front. Surg. 2017, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Maranzano, E.; Trippa, F.; Pacchiarini, D.; Chirico, L.; Basagni, M.L.; Rossi, R.; Bellavita, R.; Schiavone, C.; Italiani, M.; Muti, M. Re-irradiation of brain metastases and metastatic spinal cord compression: Clinical practice suggestions. Tumori 2005, 91, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Nieder, C.; Langendijk, J.A.; Guckenberger, M.; Grosu, A.L. Second re-irradiation: A narrative review of the available clinical data. Acta Oncol. 2018, 57, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Sahgal, A.; Ma, L.; Weinberg, V.; Gibbs, I.C.; Chao, S.; Chang, U.K.; Werner-Wasik, M.; Angelov, L.; Chang, E.L.; Sohn, M.J.; et al. Reirradiation human spinal cord tolerance for stereotactic body radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 107–116. [Google Scholar] [CrossRef]
- Andreou, D.; Ruppin, S.; Fehlberg, S.; Pink, D.; Werner, M.; Tunn, P.U. Survival and prognostic factors in chondrosarcoma: Results in 115 patients with long-term follow-up. Acta Orthop. 2011, 82, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, G.; Chen, X.; Huang, X.; Liu, M.; Pan, W.; Yan, X.; Lin, N.; Ye, Z. Predictors of the survival of patients with chondrosarcoma of bone and metastatic disease at diagnosis. J. Cancer 2019, 10, 2457–2463. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Song, J.; Chen, F.; Lin, K.; Ma, X.; Jiang, J. Does Resection of the Primary Tumor Improve Survival in Patients With Metastatic Chondrosarcoma? Clin. Orthop. Relat. Res. 2019, 477, 573–583. [Google Scholar] [CrossRef]
- Ozaki, T.; Hillmann, A.; Lindner, N.; Blasius, S.; Winkelmann, W. Metastasis of chondrosarcoma. J. Cancer Res. Clin. Oncol. 1996, 122, 625–628. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Cioffi, A.; Palmerini, E.; Piperno-Neumann, S.; Perrin, C.; Chaigneau, L.; Penel, N.; Duffaud, F.; Kurtz, J.E.; et al. Advanced chondrosarcomas: Role of chemotherapy and survival. Ann. Oncol. 2013, 24, 2916–2922. [Google Scholar] [CrossRef]
- Wyman, J.J.; Hornstein, A.M.; Meitner, P.A.; Mak, S.; Verdier, P.; Block, J.A.; Pan, J.; Terek, R.M. Multidrug resistance-1 and p-glycoprotein in human chondrosarcoma cell lines: Expression correlates with decreased intracellular doxorubicin and in vitro chemoresistance. J. Orthop. Res. 1999, 17, 935–940. [Google Scholar] [CrossRef]
- Frezza, A.M.; Cesari, M.; Baumhoer, D.; Biau, D.; Bielack, S.; Campanacci, D.A.; Casanova, J.; Esler, C.; Ferrari, S.; Funovics, P.T.; et al. Mesenchymal chondrosarcoma: Prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur. J. Cancer 2015, 51, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Bertoni, F.; Bacchini, P.; Mercuri, M.; Palmerini, E.; Ferrari, S. Mesenchymal Chondrosarcoma. An Analysis of Patients Treated at a Single Institution. Tumori J. 2007, 93, 423–427. [Google Scholar] [CrossRef]
- Dantonello, T.M.; Int-Veen, C.; Leuschner, I.; Schuck, A.; Furtwaengler, R.; Claviez, A.; Schneider, D.T.; Klingebiel, T.; Bielack, S.S.; Koscielniak, E. Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: Experiences of the CWS and COSS study groups. Cancer 2008, 112, 2424–2431. [Google Scholar] [CrossRef]
- Van Maldegem, A.M.; Bovée, J.V.; Gelderblom, H. Comprehensive analysis of published studies involving systemic treatment for chondrosarcoma of bone between 2000 and 2013. Clin. Sarcoma Res. 2014, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Dhinsa, B.S.; DeLisa, M.; Pollock, R.; Flanagan, A.M.; Whelan, J.; Gregory, J. Dedifferentiated Chondrosarcoma Demonstrating Osteosarcomatous Differentiation. Oncol. Res. Treat. 2018, 41, 456–460. [Google Scholar] [CrossRef] [Green Version]
- Monga, V.; Mani, H.; Hirbe, A.; Milhem, M. Non-Conventional Treatments for Conventional Chondrosarcoma. Cancers 2020, 12. [Google Scholar] [CrossRef]
- Van Maldegem, A.; Conley, A.P.; Rutkowski, P.; Patel, S.R.; Lugowska, I.; Desar, I.M.E.; Bovée, J.; Gelderblom, H. Outcome of First-Line Systemic Treatment for Unresectable Conventional, Dedifferentiated, Mesenchymal, and Clear Cell Chondrosarcoma. Oncologist 2019, 24, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Nooij, M.A.; Whelan, J.; Bramwell, V.H.; Taminiau, A.T.; Cannon, S.; Hogendoorn, P.C.; Pringle, J.; Uscinska, B.M.; Weeden, S.; Kirkpatrick, A.; et al. Doxorubicin and cisplatin chemotherapy in high-grade spindle cell sarcomas of the bone, other than osteosarcoma or malignant fibrous histiocytoma: A European Osteosarcoma Intergroup Study. Eur. J. Cancer 2005, 41, 225–230. [Google Scholar] [CrossRef]
- Xu, K.M.; Switchenko, J.M.; Tian, S.; Escott, C.E.; Pfister, N.T.; Gillespie, T.W.; Curran, W.J.; Khan, M.K. More effective systemic therapies are needed for chondrosarcoma: A National Cancer Data Base (NCDB) analysis. J. Clin. Oncol. 2018, 36, e23508. [Google Scholar] [CrossRef]
- Kalinski, T.; Krueger, S.; Sel, S.; Werner, K.; Ropke, M.; Roessner, A. Differential expression of VEGF-A and angiopoietins in cartilage tumors and regulation by interleukin-1beta. Cancer 2006, 106, 2028–2038. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, T.; Nishida, K.; Kawai, A.; Namba, M.; Inoue, H.; Ninomiya, Y. Human chondrosarcoma secretes vascular endothelial growth factor to induce tumor angiogenesis and stores basic fibroblast growth factor for regulation of its own growth. Int. J. Cancer 2002, 97, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, H.; Weissbach, L.; Vogel, T.; Nielsen, G.P.; Faircloth, G.T.; Shao, L.; Hornicek, F.J. Antiangiogenesis treatment combined with chemotherapy produces chondrosarcoma necrosis. Clin. Cancer Res. 2003, 9, 1211–1217. [Google Scholar] [PubMed]
- Levine, A.M.; Tulpule, A.; Quinn, D.I.; Gorospe, G., 3rd; Smith, D.L.; Hornor, L.; Boswell, W.D.; Espina, B.M.; Groshen, S.G.; Masood, R.; et al. Phase I study of antisense oligonucleotide against vascular endothelial growth factor: Decrease in plasma vascular endothelial growth factor with potential clinical efficacy. J. Clin. Oncol. 2006, 24, 1712–1719. [Google Scholar] [CrossRef]
- Klenke, F.M.; Abdollahi, A.; Bertl, E.; Gebhard, M.M.; Ewerbeck, V.; Huber, P.E.; Sckell, A. Tyrosine kinase inhibitor SU6668 represses chondrosarcoma growth via antiangiogenesis in vivo. BMC Cancer 2007, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L.; Katz, D.; Loggers, E.T.; Davidson, D.; Rodler, E.T.; Pollack, S.M. Clinical benefit of antiangiogenic therapy in advanced and metastatic chondrosarcoma. Med. Oncol. 2017, 34, 167. [Google Scholar] [CrossRef] [Green Version]
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a prospective phase 2 study of pazopanib in patients with surgically unresectable or metastatic chondrosarcoma. Cancer 2020, 126, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2018, 20, S1470–S2045. [Google Scholar] [CrossRef]
- Nurse, P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem. 2002, 3, 596–603. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Ouyang, Z.; Wang, S.; Zeng, M.; Li, Z.; Zhang, Q.; Wang, W.; Liu, T. Therapeutic effect of palbociclib in chondrosarcoma: Implication of cyclin-dependent kinase 4 as a potential target. Cell Commun. Signal. 2019, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Wang, Y.; Xie, J. The Hedgehog pathway: Role in cell differentiation, polarity and proliferation. Arch. Toxicol 2015, 89, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Jiang, T.; Guo, F.; Gong, C.; Yang, K.; Wu, Y.; Huang, X.; Cheng, W.; Xu, K. Hedgehog pathway inhibitor-4 suppresses malignant properties of chondrosarcoma cells by disturbing tumor ciliogenesis. Oncol. Rep. 2014, 32, 1622–1630. [Google Scholar] [CrossRef] [Green Version]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Infinity Pharmaceuticals, Inc. Infinity Stops Phase 2 Trials of Saridegib in Chondrosarcoma and Myelofibrosis; Press Release; Business Wire: Cambridge, MA, USA, 2012; Volume 18. [Google Scholar]
- Sun, Y.; Guo, W.; Ren, T.; Liang, W.; Zhou, W.; Lu, Q.; Jiao, G.; Yan, T. Gli1 inhibition suppressed cell growth and cell cycle progression and induced apoptosis as well as autophagy depending on ERK1/2 activity in human chondrosarcoma cells. Cell Death Dis. 2014, 5, e979. [Google Scholar] [CrossRef] [Green Version]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef]
- Polychronidou, G.; Karavasilis, V.; Pollack, S.M.; Huang, P.H.; Lee, A.; Jones, R.L. Novel therapeutic approaches in chondrosarcoma. Future Oncol. 2017, 13, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Garea, A.; Esteller, M. Histone deacetylase inhibitors: Understanding a new wave of anticancer agents. Int. J. Cancer 2004, 112, 171–178. [Google Scholar] [CrossRef]
- Sakimura, R.; Tanaka, K.; Nakatani, F.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Nakamura, T.; Matsumoto, Y.; Iwamoto, Y. Antitumor effects of histone deacetylase inhibitor on Ewing’s family tumors. Int. J. Cancer 2005, 116, 784–792. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for Treatment of Cutaneous Manifestations of Advanced Primary Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Sakimura, R.; Tanaka, K.; Yamamoto, S.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Nakamura, T.; Li, Y.; Iwamoto, Y. The Effects of Histone Deacetylase Inhibitors on the Induction of Differentiation in Chondrosarcoma Cells. Clin. Cancer Res. 2007, 13, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef]
- Sheikh, T.; Patwardhan, P.; Schwartz, G.K. Abstract 5066: Combination treatment with SAHA and 5-Azacytidine (Decitabine) induces apoptosis and suppresses tumor growth in preclinical models of chondrosarcoma. Cancer Res. 2017, 77, 5066. [Google Scholar] [CrossRef]
- Venneker, S.; Kruisselbrink, A.B.; Baranski, Z.; Palubeckaite, I.; Briaire-de Bruijn, I.H.; Oosting, J.; French, P.J.; Danen, E.H.J.; Bovée, J.V.M.G. Beyond the Influence of IDH Mutations: Exploring Epigenetic Vulnerabilities in Chondrosarcoma. Cancers 2020, 12, 3589. [Google Scholar] [CrossRef]
- Wilding, C.P.; Elms, M.L.; Judson, I.; Tan, A.C.; Jones, R.L.; Huang, P.H. The landscape of tyrosine kinase inhibitors in sarcomas: Looking beyond pazopanib. Expert Rev. Anticancer Ther. 2019, 19, 971–991. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, S.; Pantaleo, M.A.; Astolfi, A.; Dagrada, G.P.; Negri, T.; Dei Tos, A.P.; Indio, V.; Morosi, C.; Gronchi, A.; Colombo, C.; et al. Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur. J. Cancer 2014, 50, 1657–1664. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Stacchiotti, S.; Boglione, A.; Ferraresi, V.; Frustaci, S.; Comandone, A.; Casali, P.G.; Ferrari, S.; Aglietta, M. A phase 2 trial of imatinib mesylate in patients with recurrent nonresectable chondrosarcomas expressing platelet-derived growth factor receptor-α or -β. Cancer 2011, 117, 826–831. [Google Scholar] [CrossRef]
- Chiang, G.G.; Abraham, R.T. Targeting the mTOR signaling network in cancer. Trends Mol. Med. 2007, 13, 433–442. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27, S43–S51. [Google Scholar] [CrossRef] [Green Version]
- Bernstein-Molho, R.; Kollender, Y.; Issakov, J.; Bickels, J.; Dadia, S.; Flusser, G.; Meller, I.; Sagi-Eisenberg, R.; Merimsky, O. Clinical activity of mTOR inhibition in combination with cyclophosphamide in the treatment of recurrent unresectable chondrosarcomas. Cancer Chemother. Pharmacol. 2012, 70, 855–860. [Google Scholar] [CrossRef]
- Perez, J.; Decouvelaere, A.V.; Pointecouteau, T.; Pissaloux, D.; Michot, J.P.; Besse, A.; Blay, J.Y.; Dutour, A. Inhibition of chondrosarcoma growth by mTOR inhibitor in an in vivo syngeneic rat model. PLoS ONE 2012, 7, e32458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, J.E.; Stevens, J.W.; Malandra, A.E.; Fredericks, D.C.; Odgren, P.R.; Buckwalter, J.A.; Morcuende, J. Osteoclast inhibition impairs chondrosarcoma growth and bone destruction. J. Orthop. Res. 2014, 32, 1562–1571. [Google Scholar] [CrossRef]
- Moriceau, G.; Ory, B.; Gobin, B.; Verrecchia, F.; Gouin, F.; Blanchard, F.; Redini, F.; Heymann, D. Therapeutic approach of primary bone tumours by bisphosphonates. Curr. Pharm. Des. 2010, 16, 2981–2987. [Google Scholar] [CrossRef]
- Gouin, F.; Ory, B.; Rédini, F.; Heymann, D. Zoledronic acid slows down rat primary chondrosarcoma development, recurrent tumor progression after intralesional curretage and increases overall survival. Int. J. Cancer 2006, 119, 980–984. [Google Scholar] [CrossRef]
- Streitbuerger, A.; Henrichs, M.; Ahrens, H.; Lanvers-Kaminzky, C.; Gouin, F.; Gosheger, G.; Hardes, J. Cytotoxic effect of clodronate and zoledronate on the chondrosarcoma cell lines HTB-94 and CAL-78. Int. Orthop. 2011, 35, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.; Ricciotti, R.W.; Mantilla, J.; Loggers, E.T.; Pollack, S.M.; Cranmer, L.D. Response to PD1 inhibition in conventional chondrosarcoma. J. Immunother. Cancer 2018, 6, 94. [Google Scholar] [CrossRef]
- Palubeckaite, I.; Venneker, S.; Briaire-de Bruijn, I.H.; van den Akker, B.E.; Krol, A.D.; Gelderblom, H.; Bovee, J. Selection of Effective Therapies Using Three-Dimensional in vitro Modeling of Chondrosarcoma. Front. Mol. Biosci. 2020, 7, 566291. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.-C.; Hu, Y.-C. Treatment method and prognostic factors of chondrosarcoma: Based on Surveillance, Epidemiology, and End Results (SEER) database. Trans. Cancer Res. 2020, 9, 4250–4266. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lee, H.S.; Mun, Y.-H.; Koh, S.; Park, J.-S.; Lee, S.M.; Kang, N.-W.; Lee, M.Y.; Cho, C.-W.; Kim, D.-D.; et al. An overview of chondrosarcoma with a focus on nanoscale therapeutics. J. Pharm. Invest. 2020, 50, 537–552. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G. Drug Repurposing as a Source of Innovative Therapies in Chondrosarcoma. In Proceedings of the Connective Tissue Oncology Society (CTOS) 2018 Annual Meeting, Rome, Italy, 14–17 November 2018. [Google Scholar]

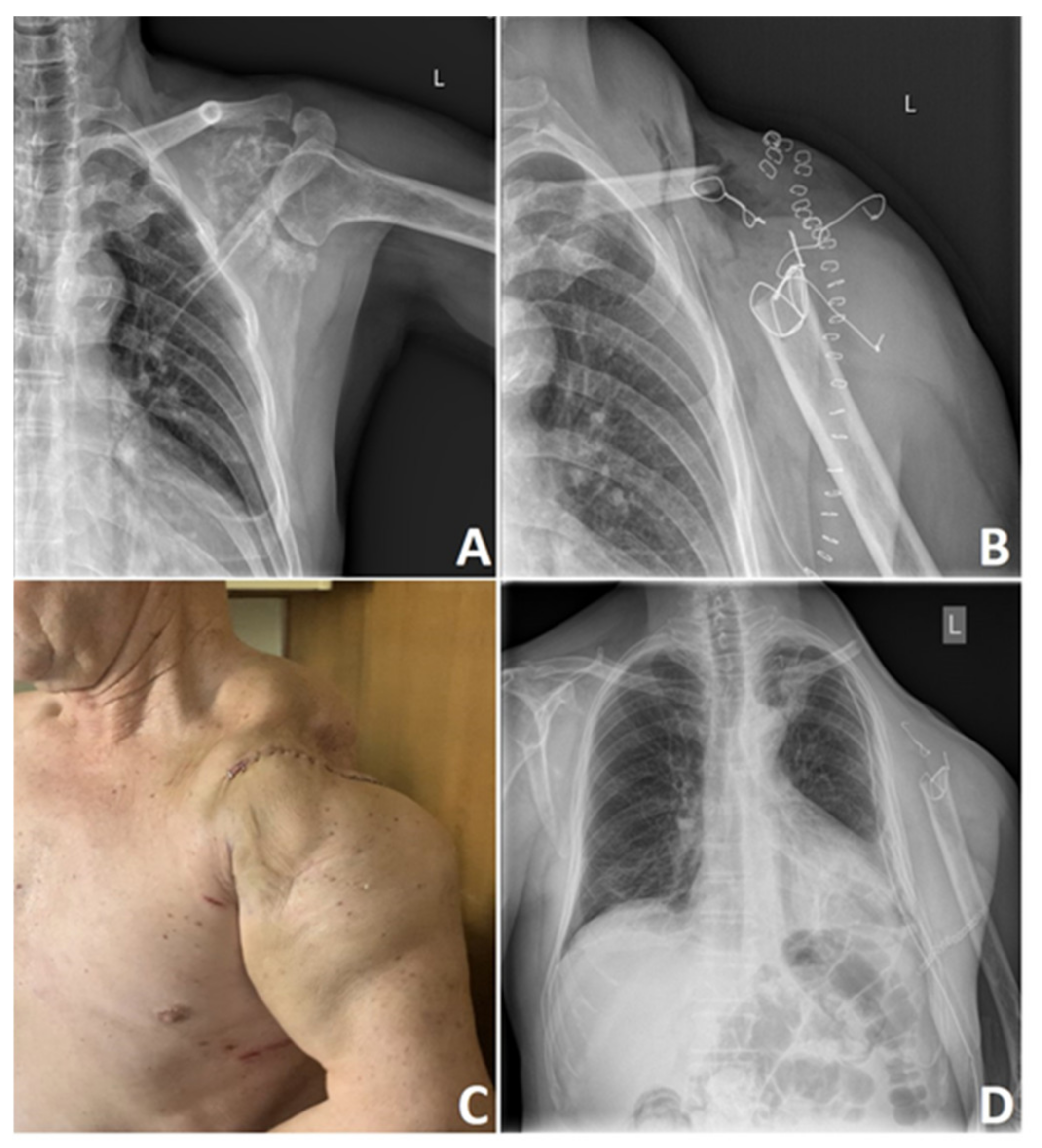
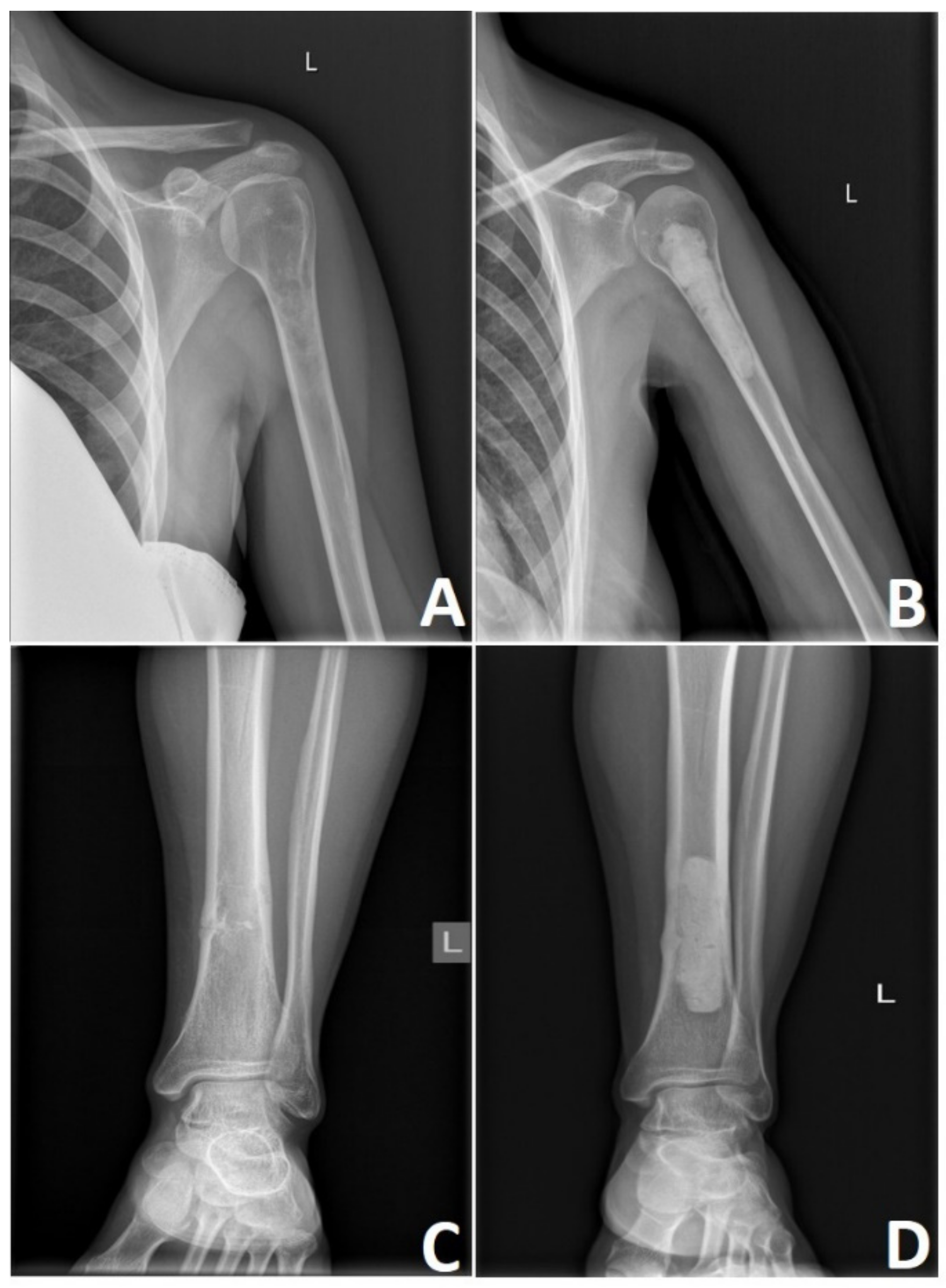


| Recommendations | Sequence | Microscopically Positive Margins | Macroscopically Positive Margins | Unresectable Tumors |
|---|---|---|---|---|
| National Comprehensive Cancer Network | perioperative | preoperative: 19.8–50.4 Gy boost: up to 70 Gy | preoperative: 19.8–50.4 Gy boost: up to 72–78 Gy | not applicable |
| postoperative | 70 Gy | >70 Gy | >70 Gy | |
| Scandinavian Sarcoma Group | postoperative | 56–62 Gy | 64–70 Gy | ≥70 Gy |
| Spanish Sarcoma Group | postoperative | >60 Gy | >60 Gy | >60 Gy 40–70 Gy * |
| Clinical Trial | Agent/ Interventions | Phase | Study Population | Status |
|---|---|---|---|---|
| NCT04278781 | AG-120 | Phase 2 | IDH1 mutant chondrosarcoma | Recruiting |
| NCT02389244 | Regorafenib | Phase 2 | Metastatic bone sarcoma, chondrosarcoma | Recruiting |
| NCT04040205 | Abemaciclib | Phase 2 | Advanced bone sarcoma, including chondrosarcoma | Recruiting |
| NCT03173976 | Zoledronic acid | Phase 1b | Resectable chondrosarcoma | Recruiting |
| NCT04340843 | Combination belinostat and guadecitabine | Phase 2 | Conventional Chondrosarcoma | Not yet recruiting |
| NCT03277924 | Nivolumab plus Sunitinib | Phase 1/2 | Advanced bone sarcomas | Recruiting |
| NCT03190174 | Combination nivolumab and nab-rapamycin (ABI-009) | Phase 1/2 | Advanced malignancies, including sarcomas with deficient mismatch repair | Active, not recruiting |
| NCT03474640 | Toripalimab | Phase 1 | Advanced malignancies including chondrosarcoma | Recruiting |
| NCT04690725 | TQB3525 | Phase 1 Phase 2 | Advanced Bone Sarcomas | Active, not recruiting |
| NCT04762602 | HMPL-306 | Phase 1 | Isocitrate Dehydrogenase Gene Mutation | Not recruiting |
| NCT03670069 | Itacitinib | Phase 1 | Metastatic Chondrosarcoma Sarcoma Tumor Immune Microenvironment | Recruiting |
| NCT03449108 | Aldesleukin Autologous Tumor-Infiltrating Lymphocytes LN-145 Autologous Tumor-Infiltrating Lymphocytes LN-145-S1 Cyclophosphamide Fludarabine | Phase 2 | Bone Sarcoma Dedifferentiated Chondrosarcoma Giant Cell Tumor of Bone Malignancy in Giant Cell Tumor of Bone Malignant Solid Neoplasm | Recruiting |
| NCT03684811 | Drug: FT-2102 Drug: Azacitidine Biological: Nivolumab Drug: Gemcitabine and Cisplatin | Phase 1 Phase 2 | Cohort 1a and 1b: Glioma Cohort 1a and 1b: Glioblastoma Multiforme Cohort 2a and 2b: Hepatobiliary Tumors (Hepatocellular Carcinoma, Bile Duct Carcinoma, Intrahepatic Cholangiocarcinoma, Other Hepatobiliary Carcinomas) Cohort 3a and 3b: Chondrosarcoma Cohort 4a and 4b: Intrahepatic Cholangiocarcinoma Cohort 5a: Other Solid Tumors With IDH1 Mutations | Recruiting |
| NCT04521686 | LY3410738 | Phase 1 | Cholangiocarcinoma Chondrosarcoma Any Solid Tumor | Recruiting |
| NCT04305548 | Trabectedin | Phase 2 | Advanced Rearranged Mesenchymal Chondrosarcoma | Not yet recruiting |
| NCT04458922 | Atezolizumab | Phase 2 | Chondrosarcoma NCI Grade 2 Chondrosarcoma NCI Grade 3 Clear Cell Sarcoma of Soft Tissue Dedifferentiated Chondrosarcoma Primary Central Chondrosarcoma | Recruiting |
| NCT01267955 | Vismodegib | Phase 2 | Clear Cell Chondrosarcoma Dedifferentiated Chondrosarcoma Locally Advanced Chondrosarcoma Mesenchymal Chondrosarcoma Metastatic Chondrosarcoma Primary Central Chondrosarcoma Unresectable Primary Central Chondrosarcoma | Active, not recruiting |
| NCT02821507 | sirolimus and cyclophosphamide | Phase 2 | Conventional Chondrosarcoma Myxoid Liposarcoma Mesenchymal Chondrosarcoma Dedifferentiated Chondrosarcoma | Recruiting |
| NCT02066285 | Pazopanib | Phase 2 | Solitary Fibrous Tumor Extraskeletal Myxoid Chondrosarcoma | Active, not recruiting |
| 2010-019817-20 | GDC-0449 | Phase 2 | Advanced chondrosarcomas | Ongoing |
| NCT04260113 | Apatinib Mesylate | Not Applicable | Unresectable Advanced Chondrosarcoma | Active, not recruiting |
| NCT02073994 | AG-120 | Phase 1 | Cholangiocarcinoma Chondrosarcoma Glioma Other Advanced Solid Tumors | Active, not recruiting |
| NCT02048371 | Regorafenib | Phase 2 | Liposarcoma Osteogenic Sarcoma Ewing’s/Ewing-like Sarcoma Rhabdomyosarcoma Mesenchymal Chondrosarcoma | Recruiting |
| NCT01182753 | Radiation: carbon ion therapy Radiation: proton therapy | Phase 3 | Chondrosarcoma | Recruiting |
| NCT02838602 | Radiation: Carbon ions therapy Radiation: Advanced external radiotherapy by Xrays or protons | Not Applicable | Malignant Tumors as Chordoma, Adenoid Cystic Carcinoma, and Sarcoma | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zając, A.E.; Kopeć, S.; Szostakowski, B.; Spałek, M.J.; Fiedorowicz, M.; Bylina, E.; Filipowicz, P.; Szumera-Ciećkiewicz, A.; Tysarowski, A.; Czarnecka, A.M.; et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers 2021, 13, 2390. https://doi.org/10.3390/cancers13102390
Zając AE, Kopeć S, Szostakowski B, Spałek MJ, Fiedorowicz M, Bylina E, Filipowicz P, Szumera-Ciećkiewicz A, Tysarowski A, Czarnecka AM, et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers. 2021; 13(10):2390. https://doi.org/10.3390/cancers13102390
Chicago/Turabian StyleZając, Agnieszka E., Sylwia Kopeć, Bartłomiej Szostakowski, Mateusz J. Spałek, Michał Fiedorowicz, Elżbieta Bylina, Paulina Filipowicz, Anna Szumera-Ciećkiewicz, Andrzej Tysarowski, Anna M. Czarnecka, and et al. 2021. "Chondrosarcoma-from Molecular Pathology to Novel Therapies" Cancers 13, no. 10: 2390. https://doi.org/10.3390/cancers13102390
APA StyleZając, A. E., Kopeć, S., Szostakowski, B., Spałek, M. J., Fiedorowicz, M., Bylina, E., Filipowicz, P., Szumera-Ciećkiewicz, A., Tysarowski, A., Czarnecka, A. M., & Rutkowski, P. (2021). Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers, 13(10), 2390. https://doi.org/10.3390/cancers13102390