
Familial Occurrence of Adult Granulosa Cell Tumors: Analysis of Whole-Genome Germline Variants


 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
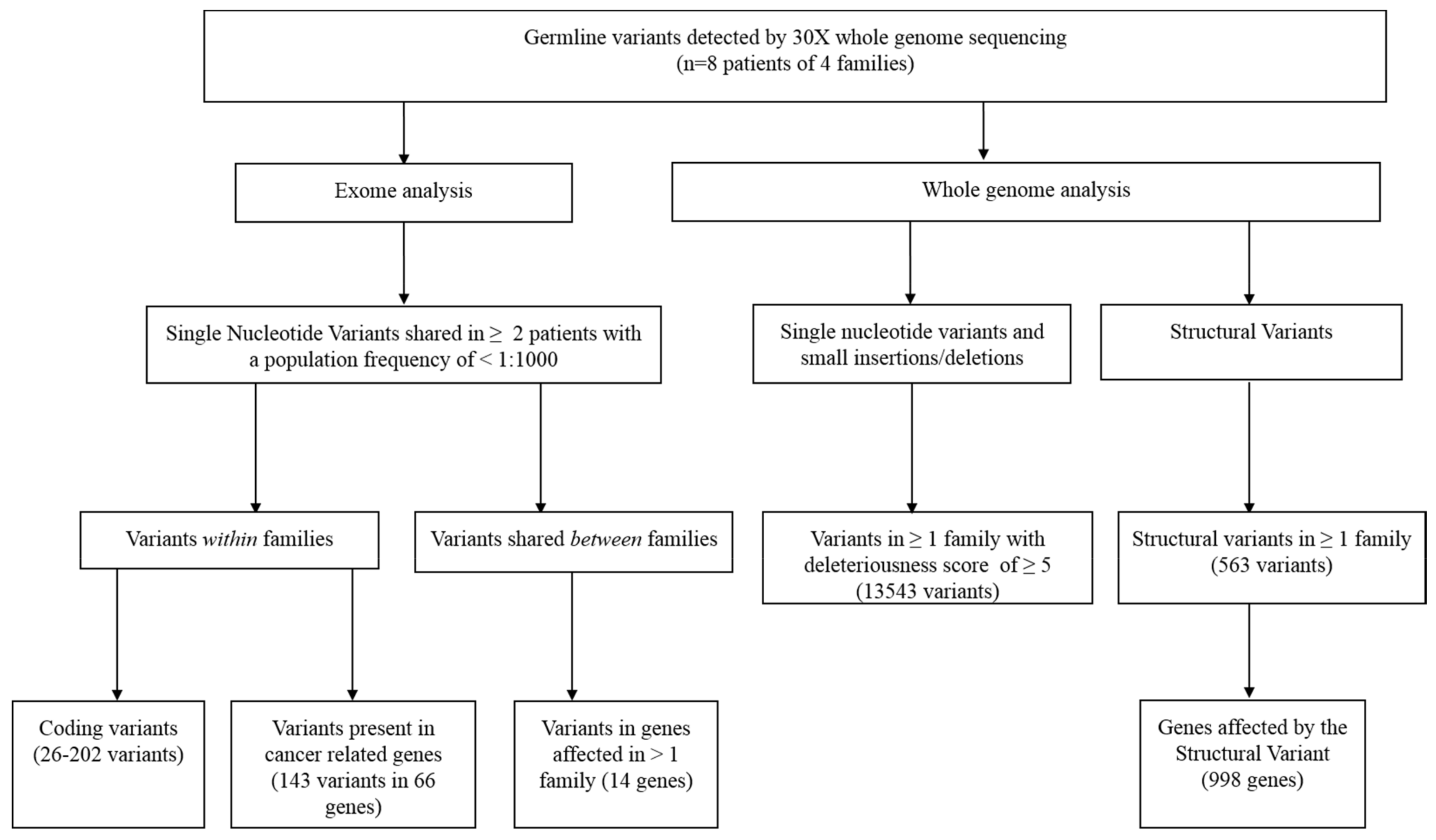
2. Materials and Methods
2.1. Whole-Genome Sequencing and Variant Calling
2.2. Genome Analysis
3. Results
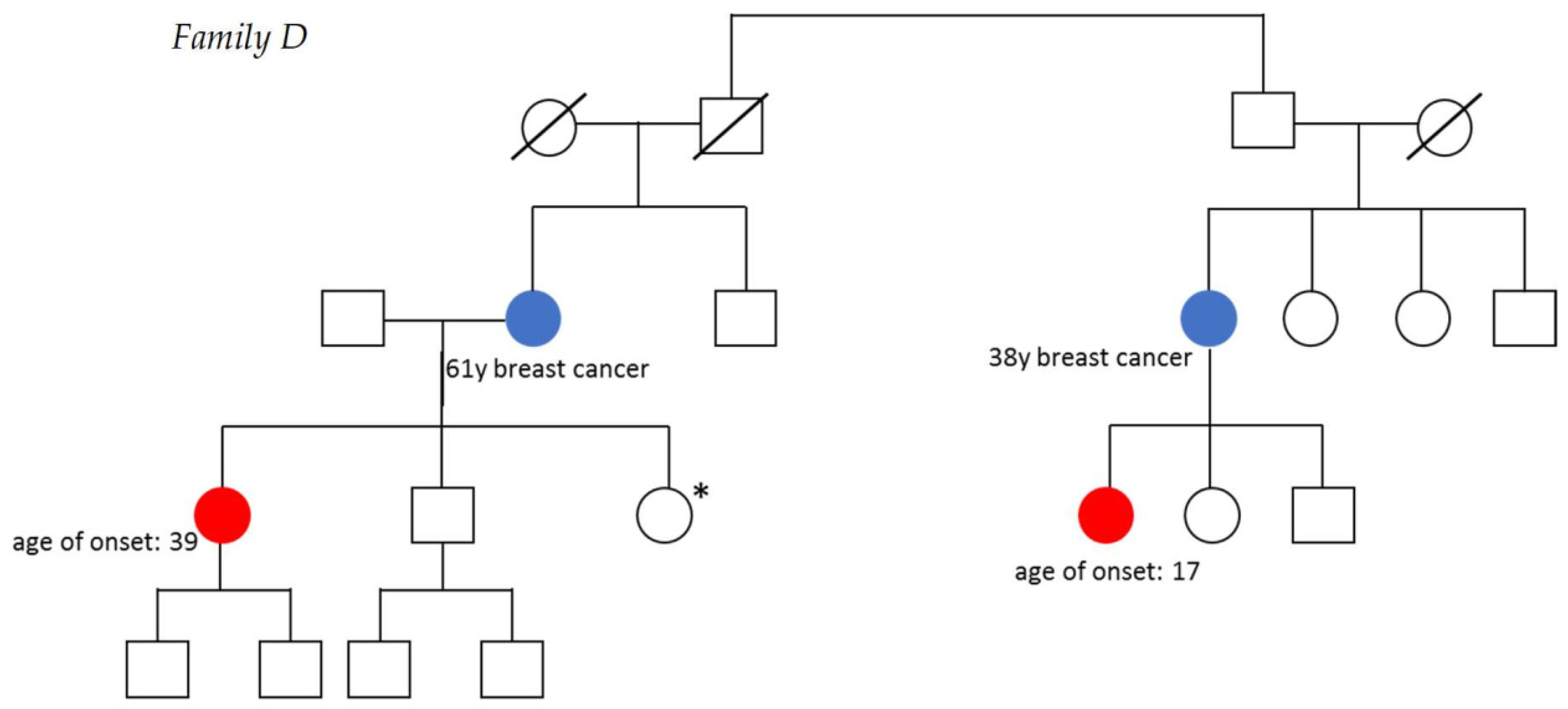
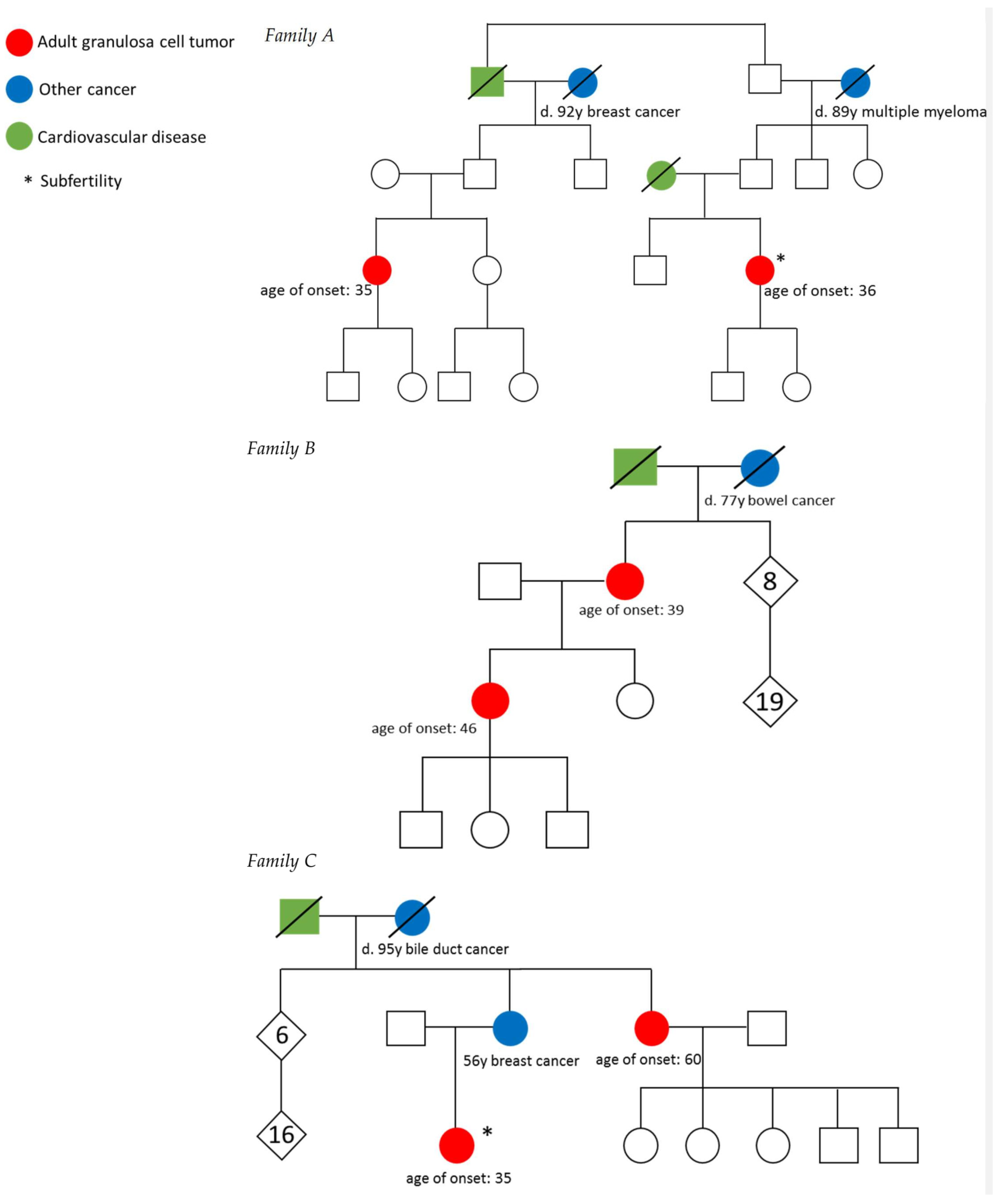
3.1. Description of Families
3.2. Exome Analysis
3.3. Whole-Genome Analysis
3.4. Variants in Genes Associated with Sex Cord-Stromal Tumor or Hereditary Ovarian Cancer
3.5. Comparison with Unrelated AGCT Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Schumer, S.T.; Cannistra, S.A. Granulosa cell tumor of the ovary. J. Clin. Oncol. 2003, 21, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Gainder, S.; Kaur, J.; Siwatch, S.; Gupta, N. Adult Granulosa Cell Tumor: A Sinister Differential for Clomiphene-resistant Infertility. J. Hum. Reprod. Sci. 2018, 11, 190–192. [Google Scholar]
- Lappohn, R.E.; Burger, H.G.; Bouma, J.; Bangah, M.; Krans, M.; de Bruijn, H.W. Inhibin as a marker for granulosa-cell tumors. N. Engl. J. Med. 1989, 321, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2016, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz-Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inheritance in Man. Available online: https://www.omim.org/entry/601200 (accessed on 2 February 2021).
- Fuller, P.J.; Leung, D.; Chu, S. Genetics and genomics of ovarian sex cord-stromal tumors. Clin. Genet. 2017, 91, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.P.; Kobel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009, 360, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Yoon, S.; Park, M.; Park, H.-O.; Ko, J.-J.; Lee, K.; Bae, J. Differential apoptotic activities of wild-type FOXL2 and the adult-type granulosa cell tumor-associated mutant FOXL2 (C134W). Oncogene 2011, 30, 1653–1663. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inheritance in Man. Available online: https://www.omim.org/entry/110100 (accessed on 2 February 2021).
- Stevens, T.A.; Brown, J.; Zander, D.S.; Bevers, M.W.; Gershenson, D.M.; Ramondetta, L.M. Adult granulosa cell tumors of the ovary in two first-degree relatives. Gynecol. Oncol. 2005, 98, 502–505. [Google Scholar] [CrossRef]
- Roze, J.; Monroe, G.; Kutzera, J.; Groeneweg, J.; Stelloo, E.; Paijens, S.; Nijman, H.; van Meurs, H.; van Lonkhuijzen, L.; Piek, J.; et al. Whole genome analysis of ovarian granulosa cell tumors reveals tumor heterogeneity and a high- grade tp53-specific subgroup. Cancers 2020, 12, 1308. [Google Scholar] [CrossRef] [PubMed]
- Roze, J.; Groeneweg, J.; Monroe, G.; Fransen, I.; Gultekin, M.; Zweemer, R.; Verheijen, R.H.M. Patient engagement in research on rare gynecological tumors. Int. J. Gynecol. Cancer 2020, ijgc-2020. [Google Scholar] [CrossRef] [PubMed]
- Twine, N.; Szul, P.; Henden, L.; McCann, E.; Blair, I.; Williams, K.; Bauer, D. TRIBES: A user-friendly pipeline for relatedness detection and disease gene discovery. bioRxiv 2019, 1–3. [Google Scholar] [CrossRef]
- Github. Available online: https://github.com/UMCUGenetics/IAP (accessed on 11 March 2021).
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Github. Available online: https://github.com/hartwigmedical/hmftools (accessed on 11 March 2021).
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Bioconductor. Available online: https://www.bioconductor.org/packages/release/bioc/html/StructuralVariantAnnotation.html (accessed on 4 February 2021).
- Evans, D.; Young, K.; Bulman, M.; Shenton, A.; Wallace, A.; Lalloo, F. Probability of BRCA1/2 mutation varies with ovarian histology: Results from screening 442 ovarian cancer families. Clin. Genet. 2008, 73, 338–345. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man. Available online: https://www.omim.org/entry/222448 (accessed on 2 February 2021).
- The Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 5 February 2021).
- Alexiadis, M.; Rowley, S.M.; Chu, S.; Leung, D.T.H.; Stewart, C.J.R.; Amarasinghe, K.C.; Campbell, I.G.; Fuller, P.J. Mutational landscape of ovarian adult granulosa cell tumors from whole exome and targeted TERT promoter sequencing. Mol. Cancer Res. 2019, 17, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2016, 18, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Qiao, J. Expression and regulation of adipocyte fatty acid binding protein in granulosa cells and its relation with clinical characteristics of polycystic ovary syndrome. Endocrine 2011, 40, 196–202. [Google Scholar] [CrossRef]
- Elis, S.; Desmarchais, A.; Maillard, V.; Uzbekova, S.; Monget, P.; Dupont, J. Cell proliferation and progesterone synthesis depend on lipid metabolism in bovine granulosa cells. Theriogenology 2015, 83, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Gan, X.; He, H.; Hu, S.; Deng, Y.; Chen, X.; Li, L.; Hu, J.; Li, L.; Wang, J. Dynamic characteristics of lipid metabolism in cultured granulosa cells from geese follicles at different developmental stages. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, F.; Lagos, J.; Céspedes, C.; Vio, C.P.; Bronfman, M.; Marzolo, M.-P. Megalin/LRP2 Expression Is Induced by Peroxisome Proliferator-Activated Receptor -Alpha and -Gamma: Implications for PPARs’ Roles in Renal Function. PLoS ONE 2011, 6, e16794. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.T.H.; Nguyen, T.; Oliver, E.M.; Matti, J.; Alexiadis, M.; Silke, J.; Jobling, T.W.; Fuller, P.J.; Chu, S. Combined PPARγ Activation and XIAP Inhibition as a Potential Therapeutic Strategy for Ovarian Granulosa Cell Tumors. Mol. Cancer Ther. 2019, 18, 364–375. [Google Scholar] [CrossRef] [Green Version]
- AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef] [Green Version]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.M.; Huang, K.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco Calvo, M.; Bolós Fernández, V.; Medina Villaamil, V.; Aparicio Gallego, G.; Díaz Prado, S.; Grande Pulido, E. Biology of BMP signalling and cancer. Clin. Transl. Oncol. 2009, 11, 126–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangas, S.A.; Li, X.; Umans, L.; Zwijsen, A.; Huylebroeck, D.; Gutierrez, C.; Wang, D.; Martin, J.F.; Jamin, S.P.; Behringer, R.R.; et al. Conditional Deletion of Smad1 and Smad5 in Somatic Cells of Male and Female Gonads Leads to Metastatic Tumor Development in Mice. Mol. Cell. Biol. 2008, 28, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Karim, M.A.; Samad, A.; Adhikari, U.K.; Kader, M.A.; Kabir, M.M.; Islam, M.A.; Hasan, M.N. A Multi-Omics Analysis of Bone Morphogenetic Protein 5 (BMP5) mRNA Expression and Clinical Prognostic Outcomes in Different Cancers Using Bioinformatics Approaches. Biomedicines 2020, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Yang, F.; He, H.; Li, Q.; Zhang, W.; Xing, J.; Zhu, Z.; Jiang, J.; Wang, H.; Zhao, X.; et al. Alteration of tumor suppressor BMP5 in sporadic colorectal cancer: A genomic and transcriptomic profiling based study. Mol. Cancer 2018, 17, 176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7, 78206–78218. [Google Scholar] [CrossRef] [Green Version]
- SEER Cancer Statistics Review (CSR) 1975-2017. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 2 February 2021).
- Nasioudis, D.; Wilson, E.; Mastroyannis, S.A.; Sisti, G.; Haggerty, A.F.; Ko, E.M.; Latif, N.A. Increased Risk of Breast and Uterine Cancer Among Women With Ovarian Granulosa Cell Tumors. Anticancer Res. 2019, 39, 4971–4975. [Google Scholar] [CrossRef]
- Hammer, A.; Lauszus, F.F.; Petersen, A.C. Ovarian granulosa cell tumor and increased risk of breast cancer. Acta Obstet. Gynecol. Scand. 2013, 92, 1422–1425. [Google Scholar] [CrossRef]
- Bryk, S.; Pukkala, E.; Färkkilä, A.; Heikinheimo, M.; Unkila-Kallio, L.; Riska, A. Other Primary Malignancies Among Women With Adult-Type Ovarian Granulosa Cell Tumors. Int. J. Gynecol. Cancer 2018, 28, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Meisel, J.L.; Hyman, D.M.; Jotwani, A.; Zhou, Q.; Abu-Rustum, N.R.; Iasonos, A.; Pike, M.C.; Aghajanian, C. The role of systemic chemotherapy in the management of granulosa cell tumors. Gynecol. Oncol. 2015, 136, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unkila-Kallio, L.; Tiitinen, A.; Wahlström, T.; Lehtovirta, P.; Leminen, A. Reproductive features in women developing ovarian granulosa cell tumour at a fertile age. Hum. Reprod. 2000, 15, 589–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, D.N.; Zethoven, M.; McInerny, S.; Morgan, J.A.; Rowley, S.M.; Lee, J.E.A.; Li, N.; Gorringe, K.L.; James, P.A.; Campbell, I.G. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat. Commun. 2020, 11, 1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Diagnosis | Tumor Stage | Treatment ** | Medical History | Family History |
|---|---|---|---|---|---|
| A1 | 35 | IIB * | Surgery | Clubfoot | |
| A2 | 36 | IA | Surgery | Polycystic ovary syndrome, 2x vaginal delivery after in vitro fertilization | |
| B1 | 46 | IC | Surgery | ||
| B2 | 39 | Unknown | Surgery and radiotherapy | ||
| C1 | 35 | IA | Surgery | Polycystic ovary syndrome, subfertility | Breast cancer, PCOS, subfertility |
| C2 | 60 | IA | Surgery | ||
| D1 | 17 | IC | Surgery and chemotherapy | Breast cancer, subfertility | |
| D2 | 39 | IC | Surgery and chemotherapy |
| Family | Gene | Effect | Variant (cDNA) | Variant (Protein) |
|---|---|---|---|---|
| A | HTRA4 | Nonsynonymous | c.1009G > C | p.V337L |
| LRP2 | Nonsynonymous | c.2688C > G | p.H896Q | |
| PCSK9 | Nonsynonymous | c.479G > A | p.R160Q | |
| B | BMP5 | Nonsynonymous | c.1291T > C | p.Y431H |
| CRLF2 | Frameshift | c.496_497delGT | p.N166Yfs*111 | |
| FSCN3 | Nonsynonymous | c.212G > A | p.G71D | |
| HFM1 | Nonsynonymous | c.4283T > C | p.L1428S | |
| MET | Nonsynonymous | c.3409G > A | p.G1137R | |
| NOX5 | Nonsynonymous | c.1600G > C | p.G534R | |
| SPTBN5 | Nonsynonymous | c.10672T > C | p.W3558R | |
| TEAD2 | Frameshift | c.1286_1287delAT | p.Y429Cfs*55 | |
| C | CBX8 | Nonsynonymous | c.916C > T | p.R306W |
| HYDIN | Frameshift | c.6584_6585ins59 | p.P2196Ifs*17 | |
| IGSF1 | Nonsynonymous | c.709C > G | p.P237A | |
| LAMA3 | Nonsynonymous | c.701T > A | p.I234K | |
| PSMD5 | Nonsynonymous | c.820G > A | p.V274M | |
| PXDN | Nonsynonymous | c.3464C > A | p.A1155E | |
| TBP | Frameshift | c.231_234delGCAGinsCAG | p.Q77Hfs*67 | |
| D | USP44 | Nonsynonymous | c.1250G > A | p.R417H |
| RASSF2* | Nonsynonymous | c.389T > A | p.L130Q |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roze, J.F.; Kutzera, J.; Koole, W.; Ausems, M.G.E.M.; Engelstad, K.; Piek, J.M.J.; de Kroon, C.D.; Verheijen, R.H.M.; van Haaften, G.; Zweemer, R.P.; et al. Familial Occurrence of Adult Granulosa Cell Tumors: Analysis of Whole-Genome Germline Variants. Cancers 2021, 13, 2430. https://doi.org/10.3390/cancers13102430
Roze JF, Kutzera J, Koole W, Ausems MGEM, Engelstad K, Piek JMJ, de Kroon CD, Verheijen RHM, van Haaften G, Zweemer RP, et al. Familial Occurrence of Adult Granulosa Cell Tumors: Analysis of Whole-Genome Germline Variants. Cancers. 2021; 13(10):2430. https://doi.org/10.3390/cancers13102430
Chicago/Turabian StyleRoze, Joline F., Joachim Kutzera, Wouter Koole, Margreet G. E. M. Ausems, Kristi Engelstad, Jurgen M. J. Piek, Cor D. de Kroon, René H. M. Verheijen, Gijs van Haaften, Ronald P. Zweemer, and et al. 2021. "Familial Occurrence of Adult Granulosa Cell Tumors: Analysis of Whole-Genome Germline Variants" Cancers 13, no. 10: 2430. https://doi.org/10.3390/cancers13102430
APA StyleRoze, J. F., Kutzera, J., Koole, W., Ausems, M. G. E. M., Engelstad, K., Piek, J. M. J., de Kroon, C. D., Verheijen, R. H. M., van Haaften, G., Zweemer, R. P., & Monroe, G. R. (2021). Familial Occurrence of Adult Granulosa Cell Tumors: Analysis of Whole-Genome Germline Variants. Cancers, 13(10), 2430. https://doi.org/10.3390/cancers13102430