
Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathogenesis of HBV-Related HCC
2.1. Chronic Inflammation and Hepatocyte Regeneration
2.2. HBV Genotypes and Specific HBV Variants
2.3. HBx: A Multifunctional Viral Protein with Versatile Oncogenic Activities
2.3.1. HBx Stimulates HBV Gene Expression
2.3.2. HBx Effects on Tumor-Related Characteristics
2.3.3. HBx Mutants and HCC Development
2.4. Sex Hormones in Regulating the Gender Difference of Carcinogenesis
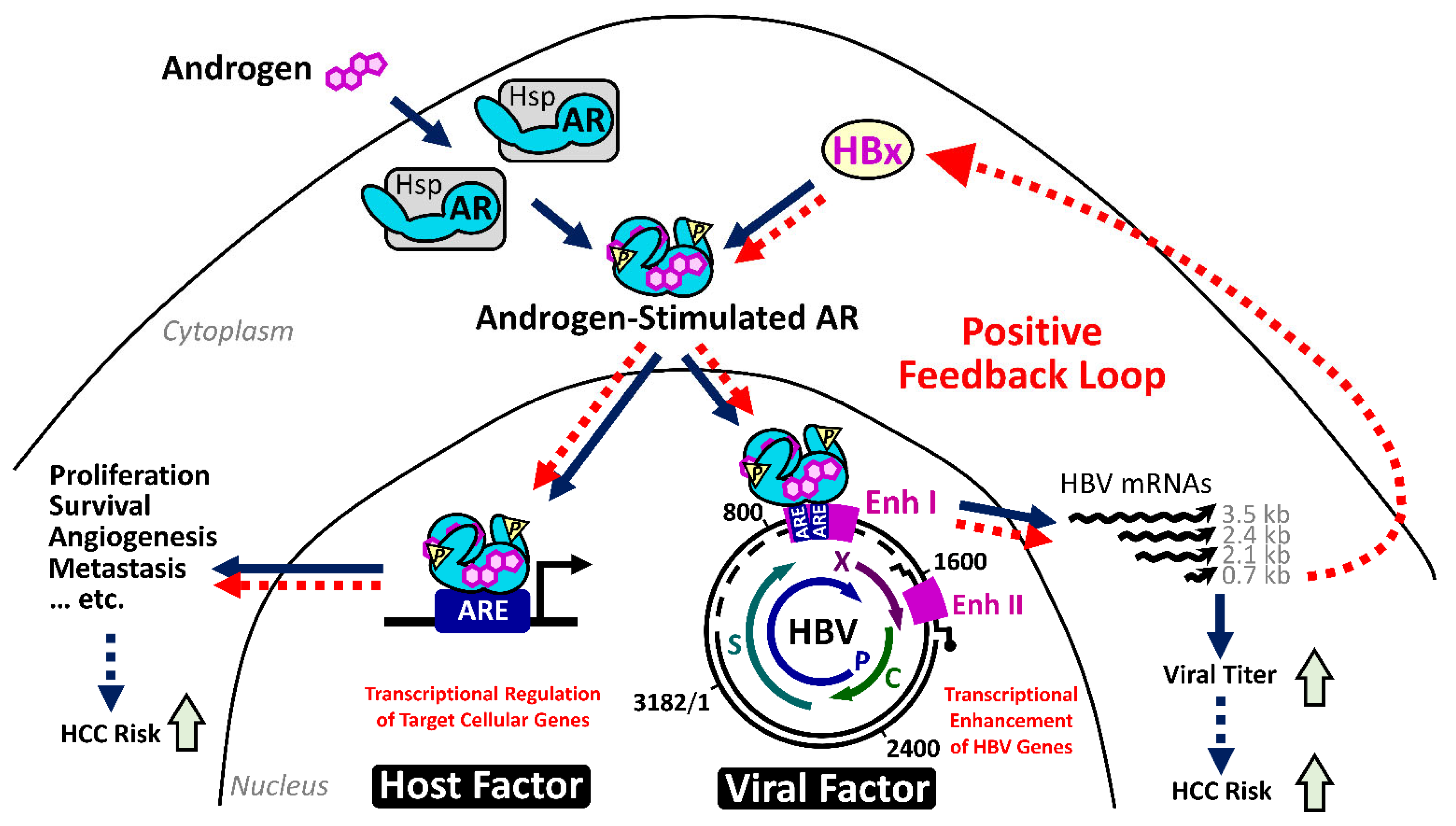
2.4.1. The HBx–AR Circuit in Promoting Male HCC
2.4.2. Estrogen/Estrogen Receptor Pathway in Suppressing Female HCC
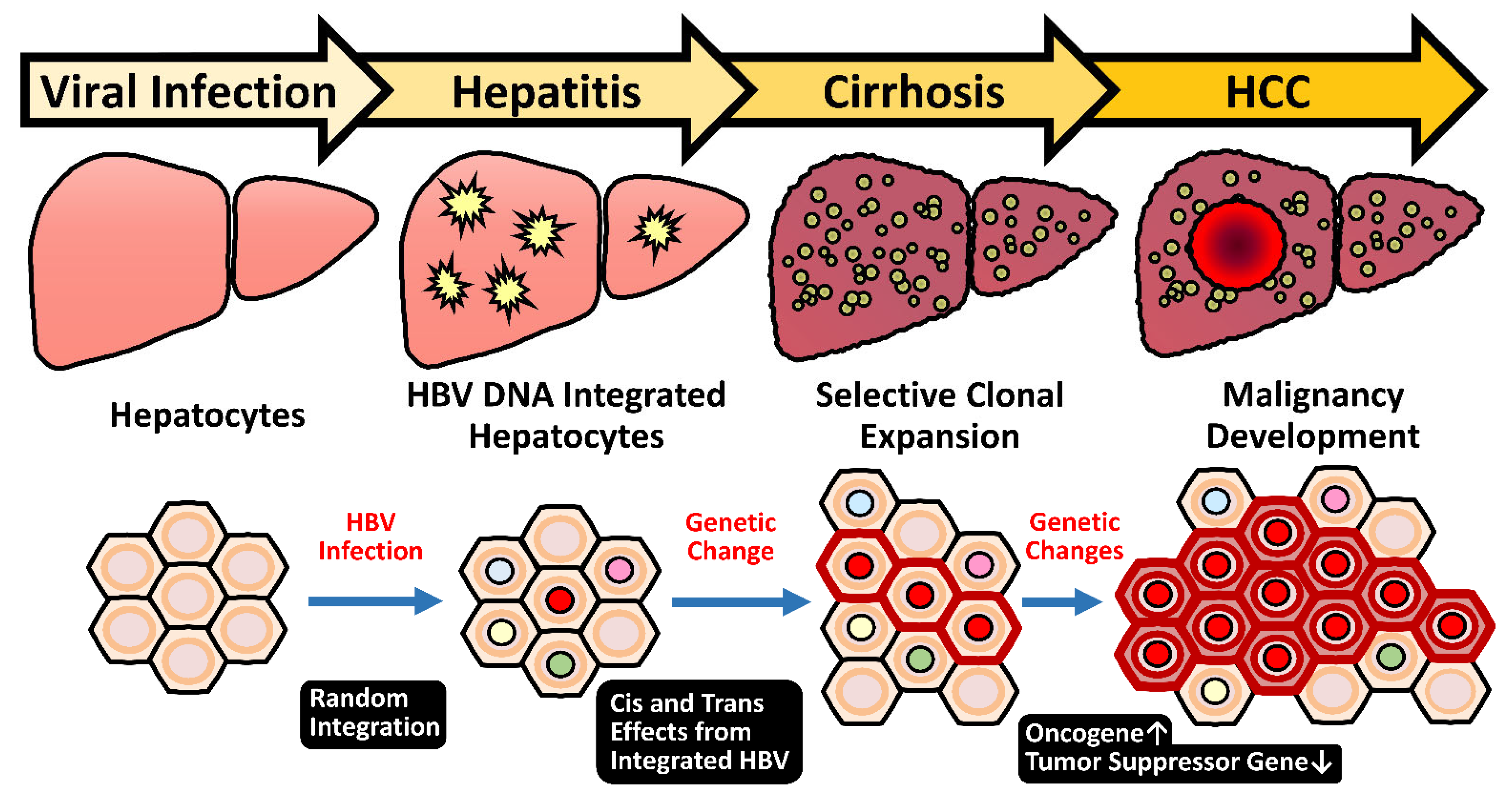
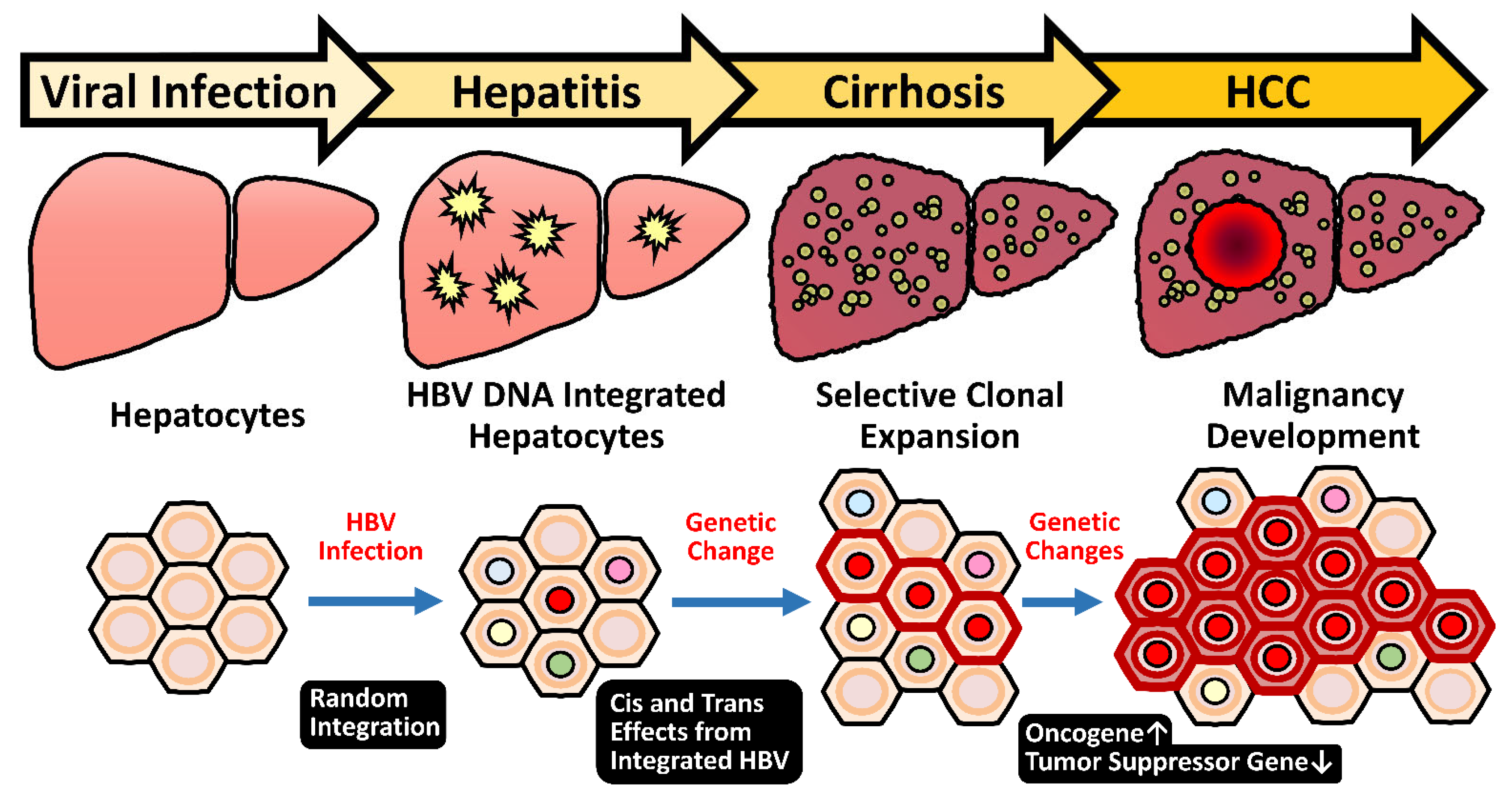
2.5. HBV Integration Induced Mutagenesis and Genomic Instability
2.5.1. Random Integration of HBV with Selective Hotspots in HCC Genome
2.5.2. HBV-Induced Insertional Mutagenesis Is Responsive to Sex Hormone Regulation
2.5.3. Genome Instability Caused by HBV Integration
2.6. Heavy Alcohol Consumption and Risk of HBV-Related Disease Progression
3. Diagnosis of HBV-Related HCC by Circulating Virus–Host Chimera Tumor DNA Generated by HBV Integration
4. The Impact of Emerging COVID-19 Pandemic on CHB Patients
5. Treatment of HBV-Related HCC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular carcinoma survival by etiology: A SEER-medicare database analysis. Hepatol. Commun. 2020, 4, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A. Epidemiology of hepatitis B virus (HBV) and hepatitis C virus (HCV) related hepatocellular carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.H.; Chung, Y.H.; Lee, H.S. Impacts of vaccination on hepatitis B viral infections in Korea over a 25-year period. Intervirology 2010, 53, 20–28. [Google Scholar] [CrossRef]
- Luo, Z.; Li, L.; Ruan, B. Impact of the implementation of a vaccination strategy on hepatitis B virus infections in China over a 20-year period. Int. J. Infect. Dis. 2012, 16, e82–e88. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Hsu, N.H.; Sung, J.L.; Hsu, T.C.; Hsu, S.T.; Kuo, Y.T.; Lo, K.J.; Shih, Y.T. A mass vaccination program in Taiwan against hepatitis B virus infection in infants of hepatitis B surface antigen-carrier mothers. JAMA J. Am. Med. Assoc. 1987, 257, 2597–2603. [Google Scholar] [CrossRef]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.H.; Chen, D.S. Hepatitis B vaccination in children: The Taiwan experience. Pathol. Biol. 2010, 58, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Polaris Observatory. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef]
- Liaw, Y.F. Hepatitis B virus replication and liver disease progression: The impact of antiviral therapy. Antivir. Ther. 2006, 11, 669–679. [Google Scholar]
- Caviglia, G.P.; Abate, M.L.; Pellicano, R.; Smedile, A. Chronic hepatitis B therapy: Available drugs and treatment guidelines. Minerva Gastroenterol. Dietol. 2015, 61, 61–70. [Google Scholar] [PubMed]
- Choi, W.M.; Choi, J.; Lim, Y.S. Effects of tenofovir vs. entecavir on risk of hepatocellular carcinoma in patients with chronic HBV infection: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2021, 19, 246–258.e9. [Google Scholar] [CrossRef] [PubMed]
- Yip, T.C.; Lai, J.C.; Wong, G.L. Secondary prevention for hepatocellular carcinoma in patients with chronic hepatitis B: Are all the nucleos(t)ide analogues the same? J. Gastroenterol. 2020, 55, 1023–1036. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tseng, C.M.; Wu, J.L.; Hsu, Y.C.; El-Serag, H.B. Magnitude of and prediction for risk of hepatocellular carcinoma in patients with chronic hepatitis B taking entecavir or tenofovir therapy: A systematic review. J. Gastroenterol. Hepatol. 2020, 35, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Saitta, C.; Tripodi, G.; Barbera, A.; Bertuccio, A.; Smedile, A.; Ciancio, A.; Raffa, G.; Sangiovanni, A.; Navarra, G.; Raimondo, G.; et al. Hepatitis B virus (HBV) DNA integration in patients with occult HBV infection and hepatocellular carcinoma. Liver Int. 2015, 35, 2311–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B virus DNA integration occurs early in the viral life cycle in an in vitro infection model via sodium taurocholate cotransporting polypeptide-dependent uptake of enveloped virus particles. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Akcay, I.M.; Katrinli, S.; Ozdil, K.; Doganay, G.D.; Doganay, L. Host genetic factors affecting hepatitis B infection outcomes: Insights from genome-wide association studies. World J. Gastroenterol. 2018, 24, 3347–3360. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Lu, S.N.; Changchien, C.S.; Yeh, C.T.; Hsu, T.T.; Tang, J.H.; Wang, J.H.; Lin, D.Y.; Chen, C.L.; Chen, W.J. Age, gender, and local geographic variations of viral etiology of hepatocellular carcinoma in a hyperendemic area for hepatitis B virus infection. Cancer 1999, 86, 1143–1150. [Google Scholar] [CrossRef]
- Chu, C.M.; Liaw, Y.F.; Sheen, I.S.; Lin, D.Y.; Huang, M.J. Sex difference in chronic hepatitis B virus infection: An appraisal based on the status of hepatitis B antigen and antibody. Hepatology 1983, 3, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Yang, H.I.; Iloeje, U.H. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology 2009, 49, S72–S84. [Google Scholar] [CrossRef] [PubMed]
- Su, F.H.; Chen, J.D.; Cheng, S.H.; Lin, C.H.; Liu, Y.H.; Chu, F.Y. Seroprevalence of hepatitis-B infection amongst Taiwanese university students 18 years following the commencement of a national hepatitis-B vaccination program. J. Med. Virol. 2007, 79, 138–143. [Google Scholar] [CrossRef]
- Liu, C.J.; Chen, P.J. Elimination of hepatitis B in highly endemic settings: Lessons learned in Taiwan and challenges ahead. Viruses 2020, 12, 815. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100. [Google Scholar] [CrossRef]
- Wong, G.L.; Lampertico, P. Residual risk of HCC during long-term oral nucleos(t)ide analogues (NUCs) in patients with CHB —Is one NUC better than the other? J. Hepatol. 2019, 71, 453–455. [Google Scholar] [CrossRef] [Green Version]
- Yip, T.C.; Wong, V.W.; Chan, H.L.; Tse, Y.K.; Lui, G.C.; Wong, G.L. Tenofovir is associated with lower risk of hepatocellular carcinoma than entecavir in patients with chronic HBV infection in China. Gastroenterology 2020, 158, 215–225.e6. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Yin, W.; Sun, R.; Wei, H.; Tian, Z. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology 2014, 59, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis B core antigen-TLR2 interactions in mice. J. Immunol. 2015, 195, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhu, Y.O.; Becht, E.; Aw, P.; Chen, J.; Poidinger, M.; Sessions, P.F.D.; Hibberd, M.L.; Bertoletti, A.; Lim, S.G.; et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Reignat, S.; Webster, G.J.; Brown, D.; Ogg, G.S.; King, A.; Seneviratne, S.L.; Dusheiko, G.; Williams, R.; Maini, M.K.; Bertoletti, A. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J. Exp. Med. 2002, 195, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Hoogeveen, R.C.; Robidoux, M.P.; Schwarz, T.; Heydmann, L.; Cheney, J.A.; Kvistad, D.; Aneja, J.; Melgaco, J.G.; Fernandes, C.A.; Chung, R.T.; et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut 2019, 68, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Schuch, A.; Alizei, E.S.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, O.; Ni, Y.; Urban, S.; et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut 2019, 68, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.J.; Reignat, S.; Brown, D.; Ogg, G.S.; Jones, L.; Seneviratne, S.L.; Williams, R.; Dusheiko, G.; Bertoletti, A. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: Implications for immunotherapy. J. Virol. 2004, 78, 5707–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoletti, A.; Kennedy, P.T.F.; Durantel, D. HBV infection and HCC: The ‘dangerous liaisons’. Gut 2018, 67, 787–788. [Google Scholar] [CrossRef]
- Kim, G.A.; Lim, Y.S.; Han, S.; Choi, J.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Lee, Y.S. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut 2018, 67, 945–952. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 2016, 151, 986–998 e4. [Google Scholar] [CrossRef] [Green Version]
- Maini, M.K.; Boni, C.; Lee, C.K.; Larrubia, J.R.; Reignat, S.; Ogg, G.S.; King, A.S.; Herberg, J.; Gilson, R.; Alisa, A.; et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 2000, 191, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Kao, J.H. Natural history of acute and chronic hepatitis B: The role of HBV genotypes and mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol. Int. 2009, 3, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Clinical and virological aspects of blood donors infected with hepatitis B virus genotypes B and C. J. Clin. Microbiol. 2002, 40, 22–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Liao, L.Y.; Liu, C.J.; Chen, P.J.; Lai, M.Y.; Kao, J.H.; Chen, D.S. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J. Gastroenterol. 2002, 37, 283–287. [Google Scholar] [CrossRef]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and viral genetic variation in HBV-related hepatocellular carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally occurring hepatitis B virus mutations leading to endoplasmic reticulum stress and their contribution to the progression of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Chang, W.T.; Chang, W.W.; Wu, H.C.; Huang, W.; Lei, H.Y.; Lai, M.D.; Fausto, N.; Su, I.J. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 2005, 41, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; Liu, H.; Gao, L.; Liu, J.; Sun, Z.; Ju, Y.; Hou, N.; Guo, C.; Liang, X.; Zhang, L.; et al. Hepatitis B virus protein preS2 potentially promotes HCC development via its transcriptional activation of hTERT. Gut 2009, 58, 1528–1537. [Google Scholar] [CrossRef]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Wang, L.H.; Huang, W.; Lai, M.D.; Su, I.J. Aberrant cyclin A expression and centrosome overduplication induced by hepatitis B virus pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 2012, 33, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Tsai, J.H.; Huang, Y.J.; Chang, W.W.; Lai, M.D.; Lei, H.Y.; Huang, W. Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol. Cancer Res. 2007, 5, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.K.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.K.; Li, C.C.; Chen, H.J.; Chang, J.L.; Jeng, K.S.; Chou, C.K.; Hsu, M.T.; Tsai, T.F. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem. Biophys. Res. Commun. 2006, 340, 916–928. [Google Scholar] [CrossRef]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Y.; Chen, J.; Cheng, G.; Xue, J. Transgenic mice expressing hepatitis B virus X protein are more susceptible to carcinogen induced hepatocarcinogenesis. Exp. Mol. Pathol. 2004, 76, 44–50. [Google Scholar] [CrossRef]
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [Green Version]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [Green Version]
- Decorsiere, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Kimata, Y.; Nomaguchi, K.; Watanabe, K.; Kohno, K. Identification of a novel non-structural maintenance of chromosomes (SMC) component of the SMC5-SMC6 complex involved in DNA repair. J. Biol. Chem. 2002, 277, 21585–21591. [Google Scholar] [CrossRef] [Green Version]
- Ampatzidou, E.; Irmisch, A.; O’Connell, M.J.; Murray, J.M. Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol. 2006, 26, 9387–9401. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, G.D.; Cortes-Ledesma, F.; Ira, G.; Torres-Rosell, J.; Uhle, S.; Farmer, S.; Hwang, J.Y.; Machin, F.; Ceschia, A.; McAleenan, A.; et al. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 2006, 8, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Machin, F.; Farmer, S.; Jarmuz, A.; Eydmann, T.; Dalgaard, J.Z.; Aragon, L. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 2005, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and characterizing interplay between hepatitis B virus X protein and Smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Yi, F.; Merrill, B.J. Repression of nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol. 2006, 26, 7479–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takigawa, Y.; Brown, A.M. Wnt signaling in liver cancer. Curr. Drug Targets 2008, 9, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. The oncogenic activation of beta-catenin. Curr. Opin. Genet. Dev. 1999, 9, 15–21. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.C.; Yang, J.Y.; Lee, D.F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Wang, E.Y.; Yeh, K.H.; Jeng, Y.M.; Horng, J.H.; Wu, L.L.; Chen, Y.T.; Huang, H.C.; Hsu, C.L.; Chen, P.J.; et al. Complement C1q mediates the expansion of periportal hepatic progenitor cells in senescence-associated inflammatory liver. Proc. Natl. Acad. Sci. USA 2020, 117, 6717–6725. [Google Scholar] [CrossRef]
- Wang, E.Y.; Yeh, S.H.; Tsai, T.F.; Huang, H.P.; Jeng, Y.M.; Lin, W.H.; Chen, W.C.; Yeh, K.H.; Chen, P.J.; Chen, D.S. Depletion of beta-catenin from mature hepatocytes of mice promotes expansion of hepatic progenitor cells and tumor development. Proc. Natl. Acad. Sci. USA 2011, 108, 18384–18389. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Geng, M.; Xin, X.; Bi, L.Q.; Zhou, L.T.; Liu, X.H. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, K.H.; Lee, J.M.; Park, J.H.; Kim, H.S. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin. Vaccine Immunol. CVI 2011, 18, 914–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Chu, Y.D.; Yeh, C.T. Emergence of oncogenic-enhancing hepatitis B virus X gene mutants in patients receiving suboptimal entecavir treatment. Hepatology 2019, 69, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Quioc-Salomon, B.; Fallot, G.; Halgand, B.; Feray, C.; Buendia, M.A.; Neuveut, C. Hepatitis B virus replicating in hepatocellular carcinoma encodes HBx variants with preserved ability to antagonize restriction by Smc5/6. Antivir. Res. 2019, 172, 104618. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Trabut, J.B.; Delpuech, O.; Carnot, F.; Colombo, M.; Kremsdorf, D.; Brechot, C.; Thiers, V. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J. Hepatol. 2003, 39, 253–261. [Google Scholar] [CrossRef]
- Ma, N.F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [Green Version]
- Amaddeo, G.; Cao, Q.; Ladeiro, Y.; Imbeaud, S.; Nault, J.C.; Jaoui, D.; Mathe, Y.G.; Laurent, C.; Laurent, A.; Bioulac-Sage, P.; et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015, 64, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.W.; Chen, C.J. Hepatitis B and C viruses in the development of hepatocellular carcinoma. Crit. Rev. Oncol. Hematol. 1994, 17, 71–91. [Google Scholar] [CrossRef]
- Yu, M.W.; Yang, Y.C.; Yang, S.Y.; Cheng, S.W.; Liaw, Y.F.; Lin, S.M.; Chen, C.J. Hormonal markers and hepatitis B virus-related hepatocellular carcinoma risk: A nested case-control study among men. J. Natl. Cancer Inst. 2001, 93, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.W.; Cheng, S.W.; Lin, M.W.; Yang, S.Y.; Liaw, Y.F.; Chang, H.C.; Hsiao, T.J.; Lin, S.M.; Lee, S.D.; Chen, P.J.; et al. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J. Natl. Cancer Inst. 2000, 92, 2023–2028. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.W.; Chang, H.C.; Chang, S.C.; Liaw, Y.F.; Lin, S.M.; Liu, C.J.; Lee, S.D.; Lin, C.L.; Chen, P.J.; Lin, S.C.; et al. Role of reproductive factors in hepatocellular carcinoma: Impact on hepatitis B- and C-related risk. Hepatology 2003, 38, 1393–1400. [Google Scholar]
- Yang, W.J.; Chang, C.J.; Yeh, S.H.; Lin, W.H.; Wang, S.H.; Tsai, T.F.; Chen, D.S.; Chen, P.J. Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3β kinase pathways. Hepatology 2009, 49, 1515–1524. [Google Scholar] [CrossRef]
- Chiu, C.M.; Yeh, S.H.; Chen, P.J.; Kuo, T.J.; Chang, C.J.; Chen, P.J.; Yang, W.J.; Chen, D.S. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. USA 2007, 104, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Gao, Y.Q.; Feng, H.; Lee, Y.Y.; Li, M.S.; Tian, Y.; Go, M.Y.; Yu, D.Y.; Cheung, Y.S.; Lai, P.B.; et al. Cell cycle-related kinase mediates viral-host signalling to promote hepatitis B virus-associated hepatocarcinogenesis. Gut 2014, 63, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Wang, H.Y.; Chen, D.S.; Chen, P.J. Identification of androgen response elements in the enhancer I of hepatitis B virus: A mechanism for sex disparity in chronic hepatitis B. Hepatology 2009, 50, 1392–1402. [Google Scholar] [CrossRef]
- Wang, S.H.; Yeh, S.H.; Chen, P.J. The driving circuit of HBx and androgen receptor in HBV-related hepatocarcinogenesis. Gut 2014, 63, 1688–1689. [Google Scholar] [CrossRef]
- Chen, P.J.; Yeh, S.H.; Liu, W.H.; Lin, C.C.; Huang, H.C.; Chen, C.L.; Chen, D.S. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology 2012, 56, 632–643. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, A.S.; Tsang, D.P.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Zhao, G.J.; Ng, S.S.; Lin, M.C.; Yu, J.; et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives beta-catenin/T cell factor-dependent hepatocarcinogenesis. J. Clin. Investig. 2011, 121, 3159–3175. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Yeh, S.H.; Shiau, C.W.; Chen, K.F.; Lin, W.H.; Tsai, T.F.; Teng, Y.C.; Chen, D.S.; Chen, P.J. Sorafenib action in hepatitis B virus X-activated oncogenic androgen pathway in liver through SHP-1. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Tan, W.; Xu, B.; Dan, Y.; Zhao, W.; Deng, C.; Chen, W.; Tan, S.; Mao, Q.; Wang, Y.; et al. A cis-acting regulatory variation of the estrogen receptor alpha (ESR1) gene is associated with hepatitis B virus-related liver cirrhosis. Hum. Mutat. 2011, 32, 1128–1136. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhou, G.; Deng, G.; Xie, W.; Dong, X.; Zhang, X.; Yu, L.; Yang, H.; Yuan, X.; Zhang, H.; et al. Estrogen receptor alpha polymorphisms associated with susceptibility to hepatocellular carcinoma in hepatitis B virus carriers. Gastroenterology 2006, 130, 2001–2009. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Kohno, N.; Tamaki, K.; Shono, M.; Huang, H.W.; He, J.H.; Yao, D.F. Female hepatology: Favorable role of estrogen in chronic liver disease with hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Sumi, D.; Hayashi, T.; Matsui-Hirai, H.; Jacobs, A.T.; Ignarro, L.J.; Iguchi, A. 17beta-estradiol inhibits NADPH oxidase activity through the regulation of p47phox mRNA and protein expression in THP-1 cells. Biochim. Biophys. Acta 2003, 1640, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Yang, S.; Liu, R.; Perez, E.; Yi, K.D.; Koulen, P.; Simpkins, J.W. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004, 1008, 147–154. [Google Scholar] [CrossRef]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Yeh, K.H.; Yuan, Q.; Xia, N.S.; Chen, D.S.; Chen, P.J. Estrogen receptor α represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4α. Gastroenterology 2012, 142, 989–998.e4. [Google Scholar] [CrossRef]
- Wang, S.H.; Chen, P.J.; Yeh, S.H. Gender disparity in chronic hepatitis B: Mechanisms of sex hormones. J. Gastroenterol. Hepatol. 2015, 23, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.H.; Yeh, S.H.; Lu, C.C.; Yu, S.L.; Chen, H.Y.; Lin, C.Y.; Chen, D.S.; Chen, P.J. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 2009, 136, 683–693. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Tokino, T. Integration of hepatitis B virus DNA and its implications for hepatocarcinogenesis. Mol. Biol. Med. 1990, 7, 243–260. [Google Scholar]
- Li, C.L.; Li, C.Y.; Lin, Y.Y.; Ho, M.C.; Chen, D.S.; Chen, P.J.; Yeh, S.H. Androgen receptor enhances hepatic telomerase reverse transcriptase gene transcription after hepatitis B virus integration or point mutation in promoter region. Hepatology 2019, 69, 498–512. [Google Scholar] [CrossRef]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, R.B.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef]
- Hessein, M.; Saad, E.G.; Mohamed, A.A.; Kamel, E.A.M.; Hady, A.M.A.; Amina, M.; Rogler, C.E. Hit-and-run mechanism of HBV-mediated progression to hepatocellular carcinoma. Tumori 2005, 91, 241–247. [Google Scholar] [CrossRef]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Ferri, S.; Castiglione, A.; Lenzi, M.; Croce, L.S.; Granito, A.; Tiribelli, C.; Bellentani, S. Natural course of chronic HCV and HBV infection and role of alcohol in the general population: The Dionysos Study. Am. J. Gastroenterol. 2008, 103, 2248–2253. [Google Scholar] [CrossRef]
- Iida-Ueno, A.; Enomoto, M.; Tamori, A.; Kawada, N. Hepatitis B virus infection and alcohol consumption. World J. Gastroenterol. 2017, 23, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.M.; Kong, C.Y.; Zhang, S.L.; Han, B.; Zhang, Z.Y.; Wang, L.S. Alcohol and HBV synergistically promote hepatic steatosis. Ann. Hepatol. 2019, 18, 913–917. [Google Scholar] [CrossRef]
- Ganesan, M.; Eikenberry, A.; Poluektova, L.Y.; Kharbanda, K.K.; Osna, N.A. Role of alcohol in pathogenesis of hepatitis B virus infection. World J. Gastroenterol. 2020, 26, 883–903. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, W.S.; Steggerda, J.; Yang, J.D.; Kuo, A.; Sundaram, V.; Lu, S.C. Current status of hepatocellular carcinoma detection: Screening strategies and novel biomarkers. Ther. Adv. Med. Oncol. 2019, 11, 1758835919869120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M. Targeted and immune therapies for hepatocellular carcinoma: Predictions for 2019 and beyond. World J. Gastroenterol. 2019, 25, 789–807. [Google Scholar] [CrossRef]
- Portolani, N.; Coniglio, A.; Ghidoni, S.; Giovanelli, M.; Benetti, A.; Tiberio, G.A.; Giulini, S.M. Early and late recurrence after liver resection for hepatocellular carcinoma: Prognostic and therapeutic implications. Ann. Surg. 2006, 243, 229–235. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American association for the study of liver diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B.; Marrero, J.A.; Rudolph, L.; Reddy, K.R. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008, 134, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Tzartzeva, K.; Obi, J.; Rich, N.E.; Parikh, N.D.; Marrero, J.A.; Yopp, A.; Waljee, A.K.; Singal, A.G. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: A meta-analysis. Gastroenterology 2018, 154, 1706–1718.e1. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.Y.; Chen, J.; Xia, C.C.; Cao, L.K.; Duan, T.; Song, B. Noninvasive imaging of hepatocellular carcinoma: From diagnosis to prognosis. World J. Gastroenterol. 2018, 24, 2348–2362. [Google Scholar] [CrossRef]
- Yang, J.D.; Kim, W.R. Surveillance for hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bolondi, L. Screening for hepatocellular carcinoma in cirrhosis. J. Hepatol. 2003, 39, 1076–1084. [Google Scholar] [CrossRef]
- Hennedige, T.; Venkatesh, S.K. Advances in computed tomography and magnetic resonance imaging of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 205–220. [Google Scholar] [CrossRef]
- Gupta, S.; Bent, S.; Kohlwes, J. Test characteristics of alpha-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C. A systematic review and critical analysis. Ann. Intern. Med. 2003, 139, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Tarao, K.; Nozaki, A.; Komatsu, H.; Komatsu, T.; Taguri, M.; Tanaka, K.; Chuma, M.; Numata, K.; Maeda, S. Real impact of tumor marker AFP and PIVKA-II in detecting very small hepatocellular carcinoma (≤2 cm, Barcelona stage 0)—Assessment with large number of cases. World J. Hepatol. 2020, 12, 1046–1054. [Google Scholar] [CrossRef]
- Loglio, A.; Iavarone, M.; Facchetti, F.; Paolo, D.D.; Perbellini, R.; Lunghi, G.; Ceriotti, F.; Galli, C.; Sandri, M.T.; Vigano, M.; et al. The combination of PIVKA-II and AFP improves the detection accuracy for HCC in HBV caucasian cirrhotics on long-term oral therapy. Liver Int. 2020, 40, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.Q.; Wang, X.Q.; Fan, G.; Wang, C.Y.; Zheng, Y.W.; Song, X.; Pan, C.C.; Chu, F.L.; Liu, Z.F.; Lu, B.R.; et al. Value of AFP and PIVKA-II in diagnosis of HBV-related hepatocellular carcinoma and prediction of vascular invasion and tumor differentiation. Infect. Agents Cancer 2020, 15, 70. [Google Scholar] [CrossRef]
- Liu, D.; Luo, Y.; Chen, L.; Chen, L.; Zuo, D.; Li, Y.; Zhang, X.; Wu, J.; Xi, Q.; Li, G.; et al. Diagnostic value of 5 serum biomarkers for hepatocellular carcinoma with different epidemiological backgrounds: A large-scale, retrospective study. Cancer Biol. Med. 2021, 18, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Xie, Q.Q.; Huang, W.; Wang, L.; Tang, S.; Fu, J. Diagnostic value of serum DNASE1L3 in hepatitis B virus-related hepatocellular carcinoma. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef]
- Li, X.; Guo, Y.; Wang, X.; Ge, A.; Wang, H.; Fan, K.; Guo, C. Clinical significance of serum miR-487b in HBV-related hepatocellular carcinoma and its potential mechanism. Infect. Dis. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, Y.; Hu, X. Identification of potential hub genes related to diagnosis and prognosis of hepatitis B virus-related hepatocellular carcinoma via integrated bioinformatics analysis. Biomed. Res. Int. 2020, 2020, 4251761. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Scholer, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.K.Y.; Costanzo, G.G.D.; Tosti, N.; Paradiso, V.; Coto-Llerena, M.; Roscigno, G.; Perrina, V.; Quintavalle, C.; Boldanova, T.; Wieland, S.; et al. Genetic profiling using plasma-derived cell-free DNA in therapy-naive hepatocellular carcinoma patients: A pilot study. Ann. Oncol. 2018, 29, 1286–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Yang, H.; Xu, H.; Wang, Y.; Ge, P.; Ren, J.; Xu, W.; Lu, X.; Sang, X.; Zhong, S.; et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget 2016, 7, 40481–40490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornesello, M.L.; Buonaguro, L.; Tatangelo, F.; Botti, G.; Izzo, F.; Buonaguro, F.M. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 2013, 102, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Ho, M.C.; Lin, Y.Y.; Tzeng, S.T.; Chen, Y.J.; Pai, H.Y.; Wang, Y.C.; Chen, C.L.; Lee, Y.H.; Chen, D.S.; et al. Cell-free virus-host chimera DNA from hepatitis B virus integration sites as a circulating biomarker of hepatocellular cancer. Hepatology 2020, 72, 2063–2076. [Google Scholar] [CrossRef]
- Rinaldi, L.; Nevola, R.; Franci, G.; Perrella, A.; Corvino, G.; Marrone, A.; Berretta, M.; Morone, M.V.; Galdiero, M.; Giordano, M.; et al. Risk of hepatocellular carcinoma after HCV clearance by direct-acting antivirals treatment predictive factors and role of epigenetics. Cancers 2020, 12, 1351. [Google Scholar] [CrossRef]
- Cabibbo, G.; Petta, S.; Barbara, M.; Missale, G.; Virdone, R.; Caturelli, E.; Piscaglia, F.; Morisco, F.; Colecchia, A.; Farinati, F.; et al. A meta-analysis of single HCV-untreated arm of studies evaluating outcomes after curative treatments of HCV-related hepatocellular carcinoma. Liver Int. 2017, 37, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridis, G.V.; Chan, H.L.; Hansen, B.E.; Janssen, H.L.; Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: Assessment and modification with current antiviral therapy. J. Hepatol. 2015, 62, 956–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Jo, C.; Lim, Y.S. Tenofovir versus entecavir on recurrence of hepatitis B virus-related hepatocellular carcinoma after Surgical resection. Hepatology 2021, 73, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Ahn, H.; Lee, D.H.; Lee, J.H.; Jung, Y.J.; Chang, Y.; Nam, J.Y.; Cho, Y.Y.; Lee, D.H.; Cho, E.J.; et al. Entecavir and tenofovir reduce hepatitis B virus-related hepatocellular carcinoma recurrence more effectively than other antivirals. J. Viral Hepat. 2018, 25, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Bucci, L.; Garuti, F.; Lenzi, B.; Pecorelli, A.; Farinati, F.; Giannini, E.G.; Granito, A.; Ciccarese, F.; Rapaccini, G.L.; Marco, M.D.; et al. The evolutionary scenario of hepatocellular carcinoma in Italy: An update. Liver Int. 2017, 37, 259–270. [Google Scholar] [CrossRef]
- Puigvehi, M.; Moctezuma-Velazquez, C.; Villanueva, A.; Llovet, J.M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Xiang, T.D.; Zheng, X. Interaction between hepatitis B virus and SARS-CoV-2 infections. World J. Gastroenterol. 2021, 27, 782–793. [Google Scholar] [CrossRef]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q.; et al. Patients with cancer appear more vulnerable to SARS-CoV-2: A multicenter study during the COVID-19 outbreak. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA J. Am. Med. Assoc. 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Pley, C.M.; McNaughton, A.L.; Matthews, P.C.; Lourenco, J. The global impact of the COVID-19 pandemic on the prevention, diagnosis and treatment of hepatitis B virus (HBV) infection. BMJ Glob. Health 2021, 6, e004275. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Guo, J.; Liu, H.; Jiang, X. Rapid detection of hepatitis B virus in blood samples using a combination of polymerase spiral reaction with nanoparticles lateral-flow biosensor. Front. Mol. Biosci. 2020, 7, 578892. [Google Scholar] [CrossRef] [PubMed]
- Vanhomwegen, J.; Kwasiborski, A.; Diop, A.; Boizeau, L.; Hoinard, D.; Vray, M.; Bercion, R.; Ndiaye, B.; Dublineau, A.; Michiyuki, S.; et al. Development and clinical validation of loop-mediated isothermal amplification (LAMP) assay to diagnose high HBV DNA levels in resource-limited settings. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sinharay, R.; Grant, A.J.; Rivett, L.; Blackwell, R.; Mells, G.; Gelson, W. Assessing efficacy of hepatocellular carcinoma prediction scores to prioritise hepatitis B surveillance in the COVID-19 era. GastroHep 2021, 3, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Q.; Li, B.; Qu, Y.; Li, Z.; Lu, L.; Li, R.; Cai, X. The diagnosis value of a novel model with 5 circulating miRNAs for liver fibrosis in patients with chronic hepatitis B. Mediat. Inflamm. 2021, 2021, 6636947. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Liu, L.X.; Chen, T.; Zhang, Y.; Zhu, X. Serum lactate level predicts 6-months mortality in patients with hepatitis B virus-related decompensated cirrhosis: A retrospective study. Epidemiol. Infect. 2021, 149, e26. [Google Scholar] [CrossRef]
- Vibert, E.; Schwartz, M.; Olthoff, K.M. Advances in resection and transplantation for hepatocellular carcinoma. J. Hepatol. 2020, 72, 262–276. [Google Scholar] [CrossRef] [Green Version]
- Kuzuya, T.; Ishigami, M.; Ito, T.; Ishizu, Y.; Honda, T.; Ishikawa, T.; Fujishiro, M. Sorafenib vs. lenvatinib as first-line therapy for advanced hepatocellular carcinoma with portal vein tumor thrombosis. Anticancer. Res. 2020, 40, 2283–2290. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, L.; Li, N.; Zheng, B.; Liu, M. Lenvatinib versus sorafenib for unresectable hepatocellular carcinoma: A cost-effectiveness analysis. J. Comp. Eff. Res. 2020, 9, 553–562. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.-H.; Yeh, S.-H.; Chen, P.-J. Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance. Cancers 2021, 13, 2454. https://doi.org/10.3390/cancers13102454
Wang S-H, Yeh S-H, Chen P-J. Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance. Cancers. 2021; 13(10):2454. https://doi.org/10.3390/cancers13102454
Chicago/Turabian StyleWang, Sheng-Han, Shiou-Hwei Yeh, and Pei-Jer Chen. 2021. "Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance" Cancers 13, no. 10: 2454. https://doi.org/10.3390/cancers13102454
APA StyleWang, S.-H., Yeh, S.-H., & Chen, P.-J. (2021). Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance. Cancers, 13(10), 2454. https://doi.org/10.3390/cancers13102454