
Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
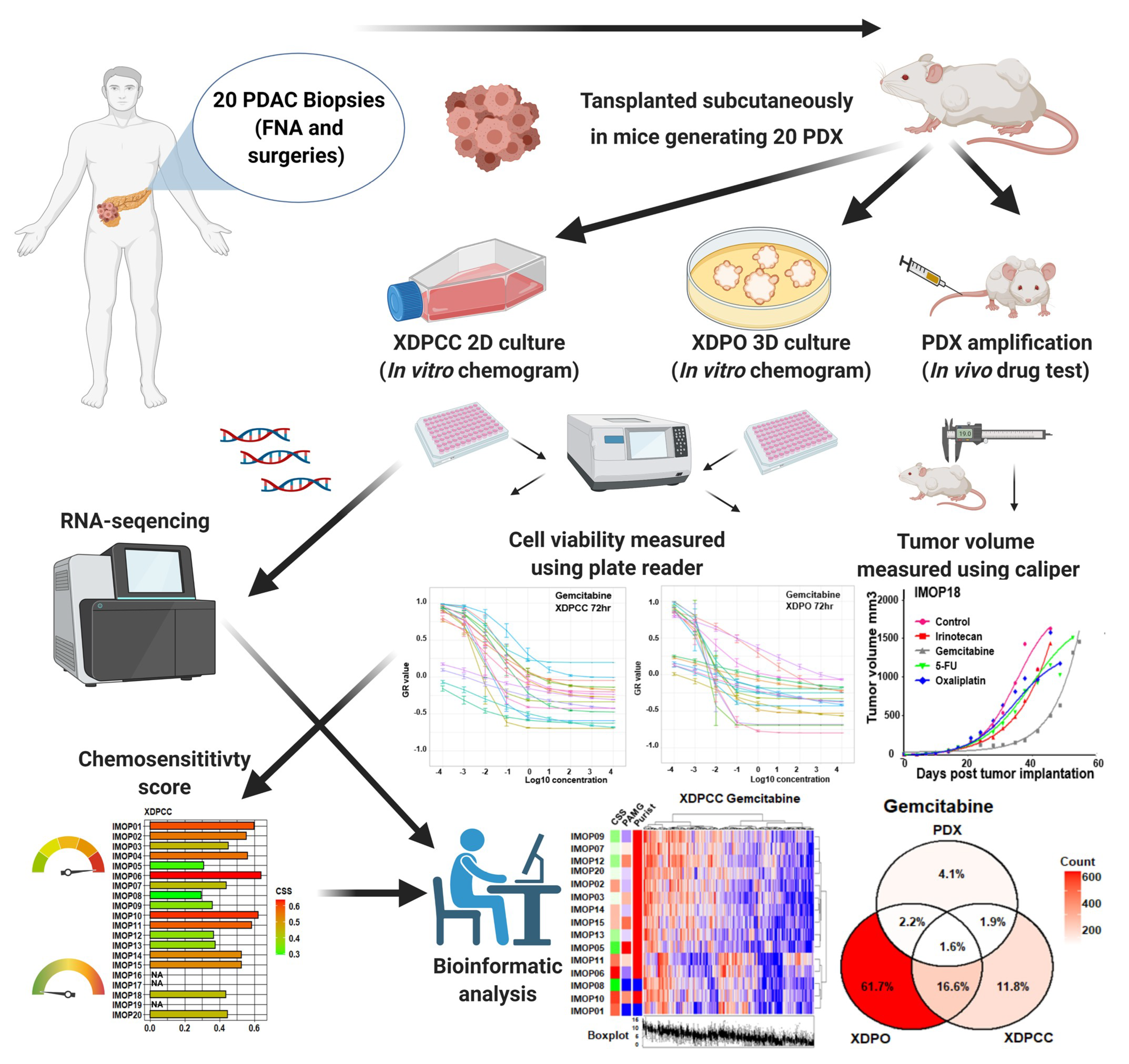
2. Materials and Methods
2.1. RNA-Sequencing Data Analysis
2.2. Establishment of PDX and In Vivo Chemosensitivity Profiling
2.3. Establishment of XDPCC and XDPO and In Vitro Chemosensitivity Profiling
2.4. Identification of Chemosensitivity Transcriptomic Profiles
2.5. Pathway and Gene Set Enrichment Analysis
3. Results
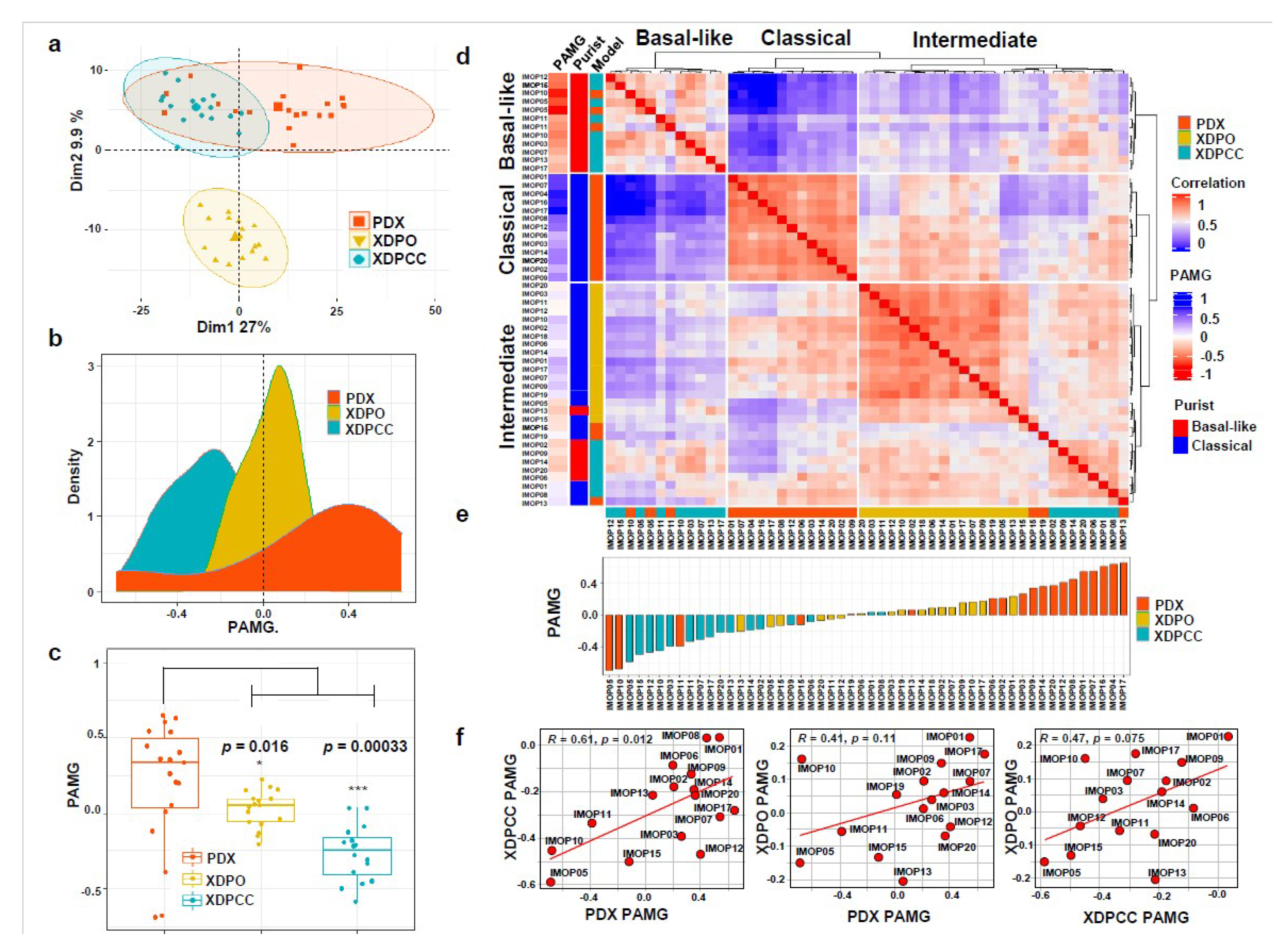
3.1. Transcriptomic Comparison between In Vitro and In Vivo Patient-Derived PDAC Preclinical Models
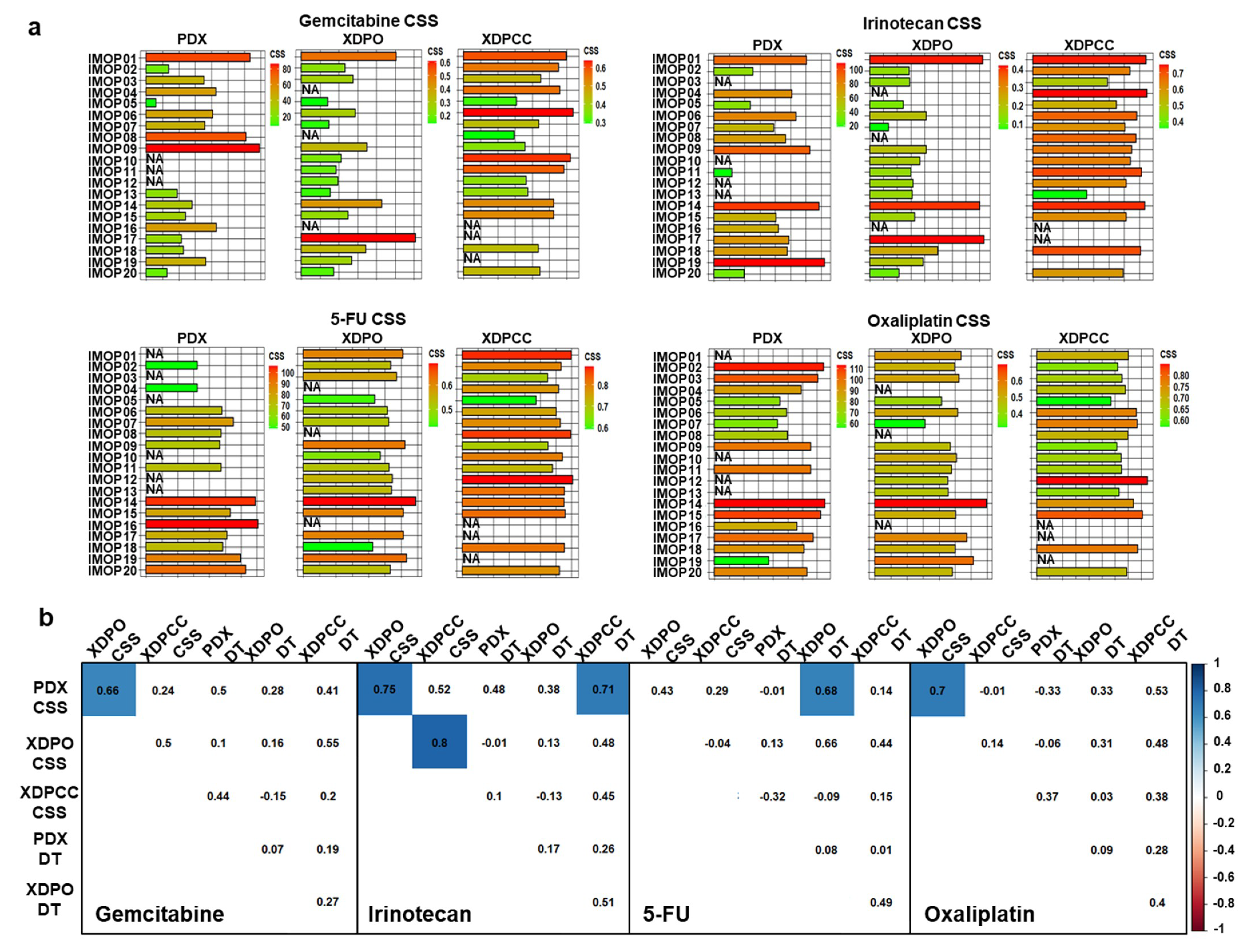
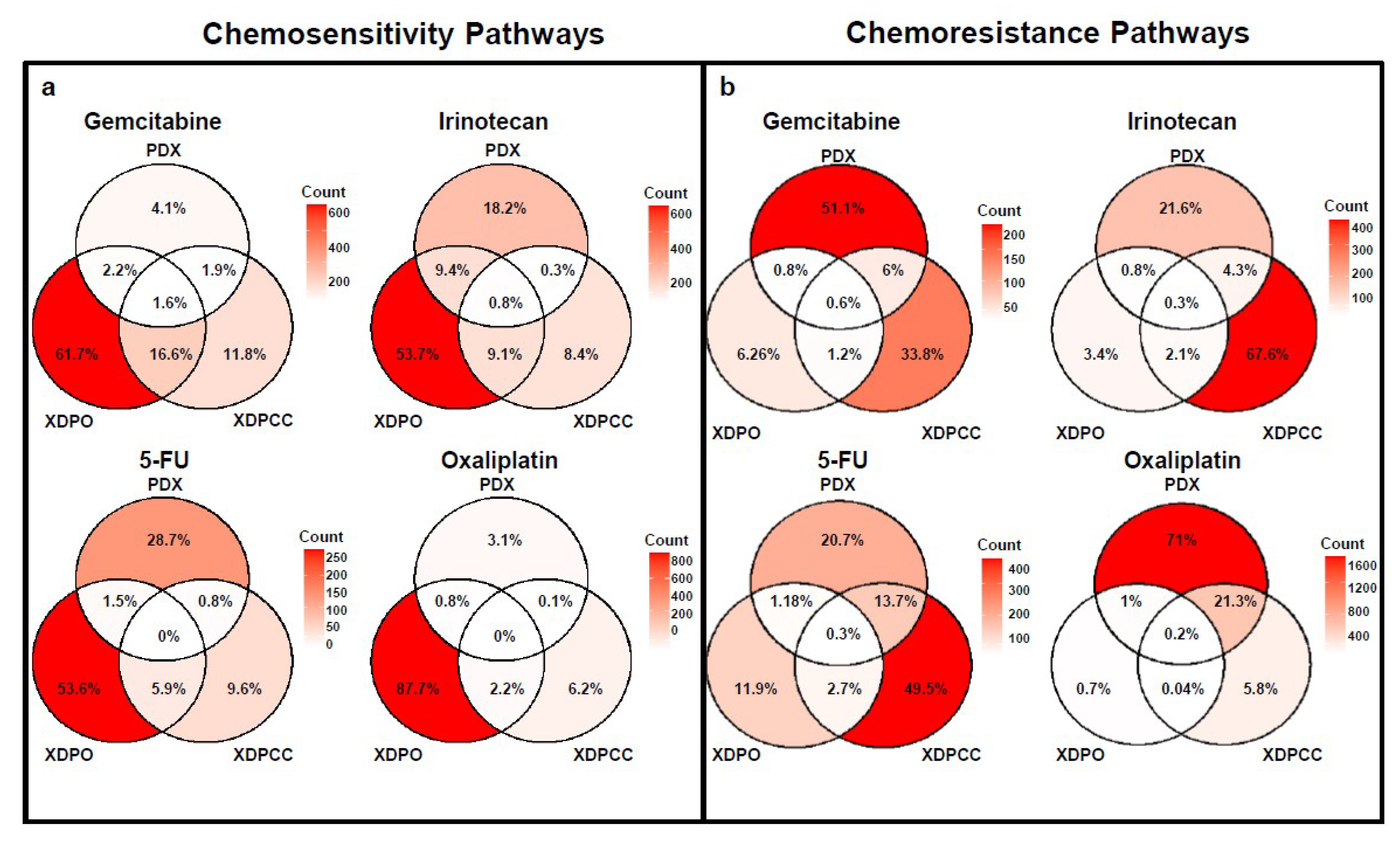
3.2. Chemosensitivity Profile Scoring and Comparison in the Different Type of Models
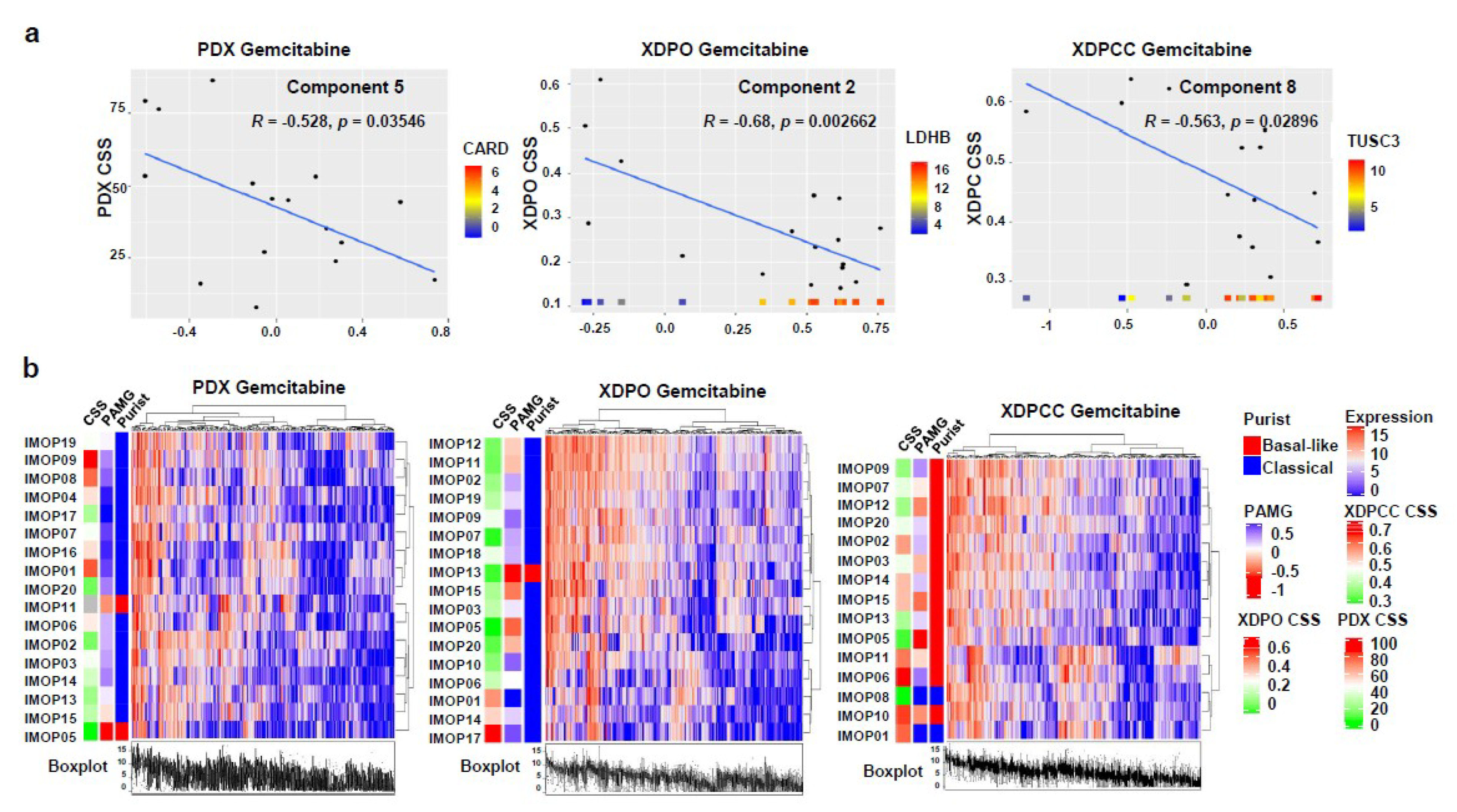
3.3. Biological Analysis of Chemosensitivity in the Different Type of Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic cancer: A growing burden. Lancet Gastroenterol. Hepatol. 2019, 12, 895–896. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Benzel, J.; Fendrich, V. Familial pancreatic cancer. Oncol. Res. Treat. 2018, 41, 611–618. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 39, 2008–2020. [Google Scholar] [CrossRef]
- Kaufmann, B.; Hartmann, D.; D’haese, J.G.; Stupakov, P.; Radenkovic, D.; Gloor, B.; Friess, H. Neoadjuvant Treatment for Borderline Resectable Pancreatic Ductal Adenocarcinoma. Dig. Surg. 2019, 36, 455–461. [Google Scholar] [CrossRef]
- Heinrich, S.; Lang, H. Neoadjuvant therapy of pancreatic cancer: Definitions and benefits. Int. J. Mol. Sci. 2017, 18, 1622. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Loeza, H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A. Specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2016, 47, 1168–1178. [Google Scholar]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [Green Version]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Iovanna, J. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, N.U.; Peng, X.L.; Jin, C.; Moffitt, R.A.; Volmar, K.E.; Belt, B.A.; Panni, R.Z.; Nywening, T.M.; Herrera, S.G.; Moore, K.J.; et al. Purity Independent Subtyping of Tumors (PurIST), A Clinically Robust, Single-sample Classifier for Tumor Subtyping in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Blum, Y.; Duconseil, P.; Vanbrugghe, C.; Brandone, N.; Poizat, F.; Roques, J.; Bigonnet, M.; Gayet, O.; Rubis, M.; et al. Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine 2020, 57, 102858. [Google Scholar] [CrossRef]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef]
- Aslan, M.; Shahbazi, R.; Ulubayram, K.; Ozpolat, B. Targeted therapies for pancreatic cancer and hurdles ahead. Anticancer Res. 2018, 38, 6591–6606. [Google Scholar] [CrossRef] [Green Version]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models—The future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef]
- Ortmann, J.; Tai, E.; Mer, A.S.; Shi, R.; Eeles, C.; Cescon, D.W.; Goldenberg, A. KuLGaP: A Selective Measure for Assessing Therapy Response in Patient-Derived Xenografts. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sereti, E.; Karagianellou, T.; Kotsoni, I.; Magouliotis, D.; Kamposioras, K.; Ulukaya, E.; Sakellaridis, N.; Zacharoulis, D.; Dimas, K. Patient Derived Xenografts (PDX) for personalized treatment of pancreatic cancer: Emerging allies in the war on a devastating cancer? J. Proteom. 2018, 188, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Yoon, K.J. Patient-derived xenograft models of pancreatic cancer: Overview and comparison with other types of models. Cancers 2020, 12, 1327. [Google Scholar] [CrossRef]
- Duconseil, P.; Gilabert, M.; Gayet, O.; Loncle, C.; Moutardier, V.; Turrini, O.; Calvo, E.; Ewald, J.; Giovannini, M.; Gasmi, M.; et al. Transcriptomic analysis predicts survival and sensitivity to anticancer drugs of patients with a pancreatic adenocarcinoma. Am. J. Pathol. 2015, 185, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Fraunhoffer, N.A.; Abuelafia, A.M.; Bigonnet, M.; Gayet, O.; Roques, J.; Telle, E.; Iovanna, J. Evidencing a Pancreatic Ductal Adenocarcinoma Subpopulation Sensitive to the Proteasome Inhibitor Carfilzomib. Clin. Cancer Res. 2020, 26, 5506–5519. [Google Scholar] [CrossRef]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. TODO: Measuring Sensitivity to Cancer Drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, 1–20. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Gandarilla, O.; Bose, A.; Muthuswamy, S.K. Pancreatic tumor organoids for modeling in vivo drug response and discovering clinically-actionable biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Frappart, P.O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Kleger, A. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolle, R.; Gayet, O.; Duconseil, P.; Vanbrugghe, C.; Roques, J.; Bigonnet, M.; Blum, Y.; Elarouci, N.; Armenoult, L.; Ayadi, M.; et al. A transcriptomic signature to predict adjuvant gemcitabine sensitivity in pancreatic adenocarcinoma. Ann. Oncol. 2021, 32, 250–260. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoare, O.; Fraunhoffer, N.; Elkaoutari, A.; Gayet, O.; Bigonnet, M.; Roques, J.; Nicolle, R.; McGuckin, C.; Forraz, N.; Sohier, E.; et al. Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models. Cancers 2021, 13, 2473. https://doi.org/10.3390/cancers13102473
Hoare O, Fraunhoffer N, Elkaoutari A, Gayet O, Bigonnet M, Roques J, Nicolle R, McGuckin C, Forraz N, Sohier E, et al. Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models. Cancers. 2021; 13(10):2473. https://doi.org/10.3390/cancers13102473
Chicago/Turabian StyleHoare, Owen, Nicolas Fraunhoffer, Abdessamad Elkaoutari, Odile Gayet, Martin Bigonnet, Julie Roques, Rémy Nicolle, Colin McGuckin, Nico Forraz, Emilie Sohier, and et al. 2021. "Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models" Cancers 13, no. 10: 2473. https://doi.org/10.3390/cancers13102473
APA StyleHoare, O., Fraunhoffer, N., Elkaoutari, A., Gayet, O., Bigonnet, M., Roques, J., Nicolle, R., McGuckin, C., Forraz, N., Sohier, E., Tonon, L., Wajda, P., Boyault, S., Attignon, V., Tabone-Eglinger, S., Barbier, S., Mignard, C., Duchamp, O., Iovanna, J., & Dusetti, N. J. (2021). Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models. Cancers, 13(10), 2473. https://doi.org/10.3390/cancers13102473