
Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma

 , and
, and 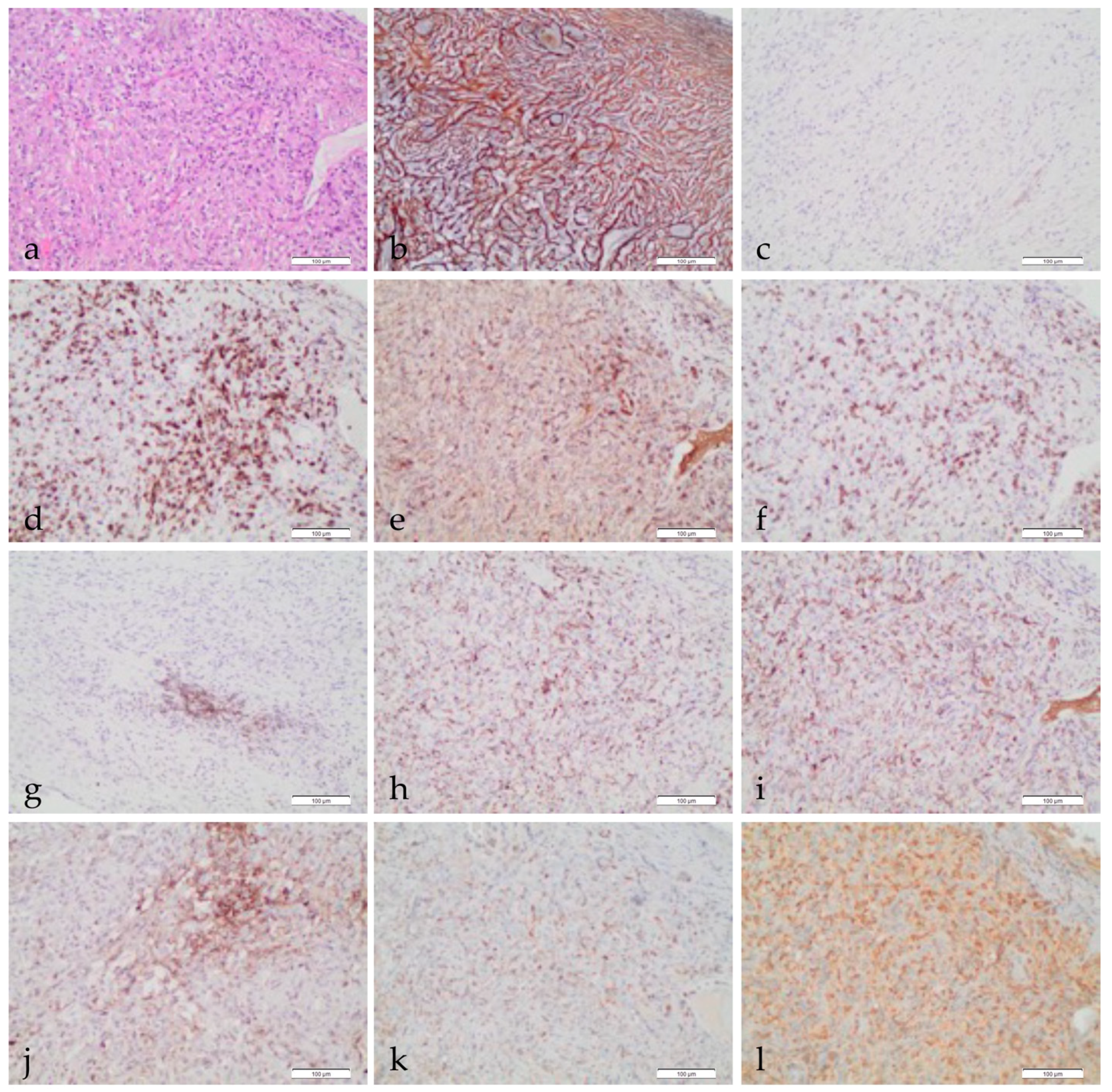{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
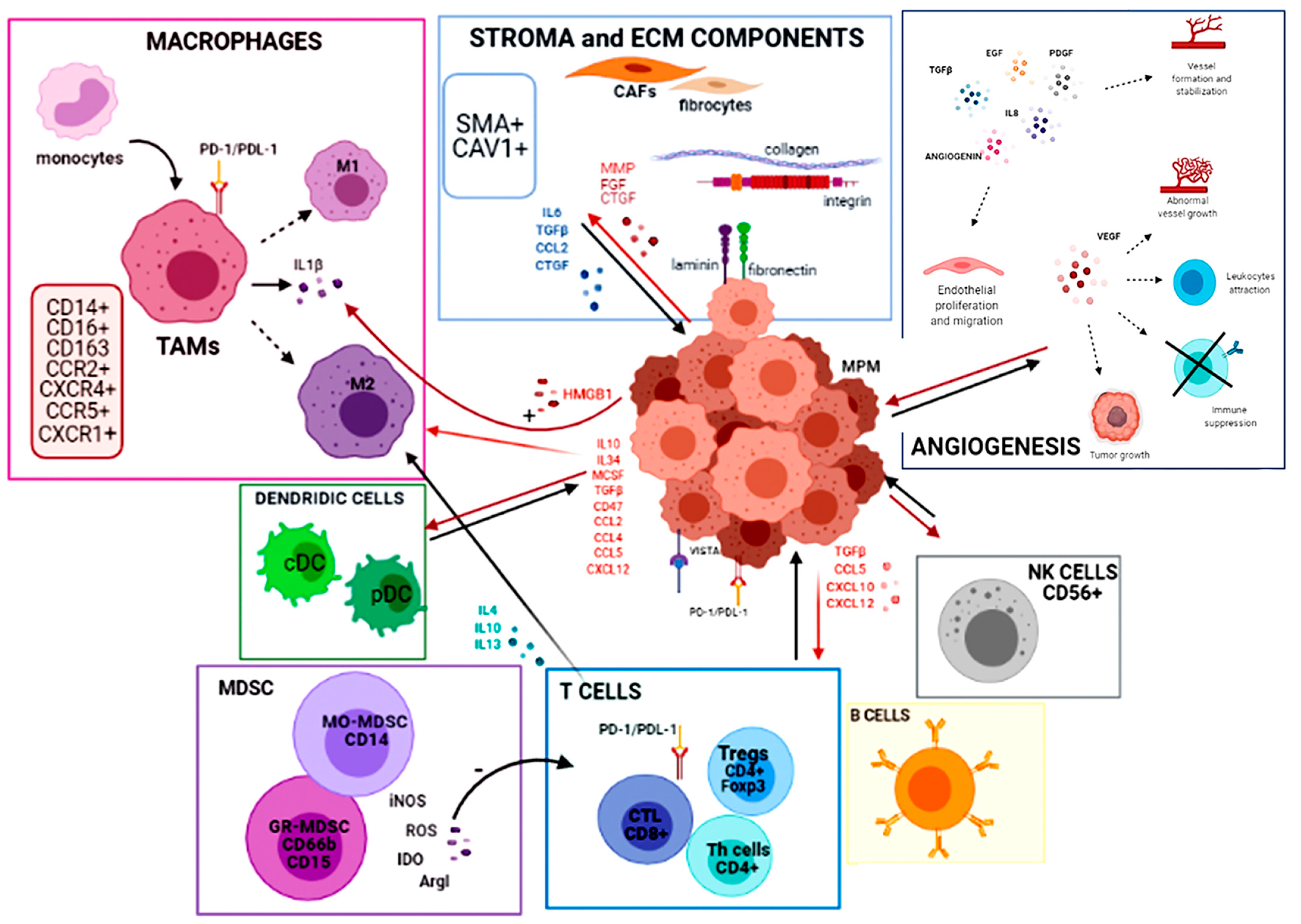
2. The Tumor Immune Microenvironment
2.1. Extracellular Matrix and Stroma Components
Cancer-Associated Fibroblasts (CAFs)
2.2. Inflammatory Cellular Component of TIME
2.2.1. Tumor-Associated Macrophages
2.2.2. T Cells and Natural Killer Cells
2.2.3. Myeloid-Derived Suppressor Cells
2.2.4. Dendritic Cells
2.2.5. B Lymphocytes
2.3. PD-L1 and Other Immune Checkpoints
3. Angiogenesis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, H.; Testa, J.R.; Carbone, M. Mesothelioma Epidemiology, Carcinogenesis, and Pathogenesis. Curr. Treat. Opt. Oncol. 2008, 9, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Kindler, H.L.; Ismaila, N.; Armato, S.G.; Bueno, R.; Hesdorffer, M.; Jahan, T.; Jones, C.M.; Miettinen, M.; Pass, H.; Rimner, A.; et al. Treatment of Malignant Pleural Mesothelioma: American Society of Clinical Oncology Clinical Practice Guideline. JCO 2018, 36, 1343–1373. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.G.; Sauter, J.L.; Nowak, A.K.; Kindler, H.L.; Gill, R.R.; Remy-Jardin, M.; Armato, S.G.; Fernandez-Cuesta, L.; Bueno, R.; Alcala, N.; et al. EURACAN/IASLC Proposals for Updating the Histologic Classification of Pleural Mesothelioma: Towards a More Multidisciplinary Approach. J. Thorac. Oncol. 2020, 15, 29–49. [Google Scholar] [CrossRef]
- Opitz, I.; Scherpereel, A.; Berghmans, T.; Psallidas, I.; Glatzer, M.; Rigau, D.; Astoul, P.; Bölükbas, S.; Boyd, J.; Coolen, J.; et al. ERS/ESTS/EACTS/ESTRO Guidelines for the Management of Malignant Pleural Mesothelioma. Eur. J. Cardio Thorac. Surg. 2020, 58, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Van Gerwen, M.; Alpert, N.; Wolf, A.; Ohri, N.; Lewis, E.; Rosenzweig, K.E.; Flores, R.; Taioli, E. Prognostic Factors of Survival in Patients with Malignant Pleural Mesothelioma: An Analysis of the National Cancer Database. Carcinogenesis 2019, 40, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.; Fennell, D.; Kerr, K.M.; Van Schil, P.E.; Haas, R.L.; Peters, S. ESMO Guidelines Committee Malignant Pleural Mesothelioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2015, 26 (Suppl. S5), v31–v39. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Cakiroglu, E.; Senturk, S. Genomics and Functional Genomics of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2020, 21, 6342. [Google Scholar] [CrossRef]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A, and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.-H.; Jen, K.-Y.; Fang, L.T.; Jones, K.; et al. Whole Exome and Targeted Deep Sequencing Identify Genome-Wide Allelic Loss and Frequent SETDB1 Mutations in Malignant Pleural Mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [Green Version]
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted Next-Generation Sequencing of Cancer Genes in Advanced Stage Malignant Pleural Mesothelioma: A Retrospective Study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The Nuclear Deubiquitinase BAP1 Is Commonly Inactivated by Somatic Mutations and 3p21.1 Losses in Malignant Pleural Mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, H.; Maeda, M.; Lee, S.; Nishimura, Y.; Kumagai-Takei, N.; Hayashi, H.; Yamamoto, S.; Hatayama, T.; Kojima, Y.; Tabata, R.; et al. Asbestos-Induced Cellular and Molecular Alteration of Immunocompetent Cells and Their Relationship with Chronic Inflammation and Carcinogenesis. J. Biomed. Biotechnol. 2012, 2012, 492608. [Google Scholar] [CrossRef] [PubMed]
- Kusamura, S.; Kepenekian, V.; Villeneuve, L.; Lurvink, R.J.; Govaerts, K.; De Hingh, I.H.J.T.; Moran, B.J.; Van der Speeten, K.; Deraco, M.; Glehen, O.; et al. Peritoneal Mesothelioma: PSOGI/EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Eur. J. Surg. Oncol. 2021, 47, 36–59. [Google Scholar] [CrossRef] [PubMed]
- Benzerdjeb, N.; Dartigues, P.; Kepenekian, V.; Valmary-Degano, S.; Mery, E.; Avérous, G.; Chevallier, A.; Laverriere, M.-H.; Villa, I.; Harou, O.; et al. Tertiary Lymphoid Structures in Epithelioid Malignant Peritoneal Mesothelioma Are Associated with Neoadjuvant Chemotherapy, but Not with Prognosis. Virchows Arch. 2021. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.; Scherpereel, A.; Nowak, A.; Fujimoto, N.; Peters, S.; Tsao, A.; Mansfield, A.; Popat, S.; Jahan, T.; Antonia, S.; et al. ID:2908 First-Line Nivolumab + Ipilimumab vs. Chemotherapy in Unresectable Malignant Pleural Mesothelioma: CheckMate 743. J. Thorac. Oncol. 2020, 15, e42. [Google Scholar] [CrossRef]
- Fennell, D.; Ottensmeier, C.; Califano, R.; Hanna, G.; Ewings, S.; Hill, K.; Wilding, S.; Danson, S.; Nye, M.; Steele, N.; et al. PS01.11 Nivolumab Versus Placebo in Relapsed Malignant Mesothelioma: The CONFIRM Phase 3 Trial. J. Thorac. Oncol. 2021, 16, S62. [Google Scholar] [CrossRef]
- Castelletti, L.; Yeo, D.; van Zandwijk, N.; Rasko, J.E.J. Anti-Mesothelin CAR T Cell Therapy for Malignant Mesothelioma. Biomark. Res. 2021, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Britschgi, C.; Schuberth, P.; Bankel, L.; Nguyen-Kim, T.D.L.; Gulati, P.; Weder, W.; Opitz, I.; Lauk, O.; Caviezel, C.; et al. Local Delivery of CAR T Cells Targeting Fibroblast Activation Protein Is Safe in Patients with Pleural Mesothelioma: First Report of FAPME, a Phase I Clinical Trial. Ann. Oncol. 2021, 32, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.P.; van Montfoort, N.; Kinderman, P.; Lukkes, M.; Klaase, L.; van Nimwegen, M.; van Gulijk, M.; Dumas, J.; Mustafa, D.A.M.; Lievense, S.L.A.; et al. Dendritic Cell Vaccination and CD40-Agonist Combination Therapy Licenses T Cell-Dependent Antitumor Immunity in a Pancreatic Carcinoma Murine Model. J. Immunother. Cancer 2020, 8, e000772. [Google Scholar] [CrossRef]
- Parra, E.R.; Zhai, J.; Tamegnon, A.; Zhou, N.; Pandurengan, R.K.; Barreto, C.; Jiang, M.; Rice, D.C.; Creasy, C.; Vaporciyan, A.A.; et al. Identification of Distinct Immune Landscapes Using an Automated Nine-Color Multiplex Immunofluorescence Staining Panel and Image Analysis in Paraffin Tumor Tissues. Sci. Rep. 2021, 11, 4530. [Google Scholar] [CrossRef] [PubMed]
- Ijsselsteijn, M.E.; van der Breggen, R.; Farina Sarasqueta, A.; Koning, F.; de Miranda, N.F.C.C. A 40-Marker Panel for High Dimensional Characterization of Cancer Immune Microenvironments by Imaging Mass Cytometry. Front. Immunol. 2019, 10, 2534. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, F.; Fernando, A.; Nica, R.; Boghiu, B.; Lungu, B.; Batra, J.; Ecker, R.C. Next-Generation Digital Histopathology of the Tumor Microenvironment. Genes 2021, 12, 538. [Google Scholar] [CrossRef]
- De Perrot, M.; Wu, L.; Cabanero, M.; Perentes, J.Y.; McKee, T.D.; Donahoe, L.; Bradbury, P.; Kohno, M.; Chan, M.-L.; Murakami, J.; et al. Prognostic Influence of Tumor Microenvironment after Hypofractionated Radiation and Surgery for Mesothelioma. J. Thorac. Cardiovasc. Surg. 2020, 159, 2082–2091.e1. [Google Scholar] [CrossRef]
- Suzuki, K.; Kadota, K.; Sima, C.S.; Sadelain, M.; Rusch, V.W.; Travis, W.D.; Adusumilli, P.S. Chronic Inflammation in Tumor Stroma Is an Independent Predictor of Prolonged Survival in Epithelioid Malignant Pleural Mesothelioma Patients. Cancer Immunol. Immunother. 2011, 60, 1721–1728. [Google Scholar] [CrossRef]
- Ujiie, H.; Kadota, K.; Nitadori, J.-I.; Aerts, J.G.; Woo, K.M.; Sima, C.S.; Travis, W.D.; Jones, D.R.; Krug, L.M.; Adusumilli, P.S. The Tumoral and Stromal Immune Microenvironment in Malignant Pleural Mesothelioma: A Comprehensive Analysis Reveals Prognostic Immune Markers. Oncoimmunology 2015, 4, e1009285. [Google Scholar] [CrossRef] [Green Version]
- Lievense, L.A.; Bezemer, K.; Cornelissen, R.; Kaijen-Lambers, M.E.H.; Hegmans, J.P.J.J.; Aerts, J.G.J.V. Precision Immunotherapy; Dynamics in the Cellular Profile of Pleural Effusions in Malignant Mesothelioma Patients. Lung Cancer 2017, 107, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring Tumour Purity and Stromal and Immune Cell Admixture from Expression Data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cheng, L.; Fan, Y.; Mao, W. Tumor Microenvironment-Associated Immune-Related Genes for the Prognosis of Malignant Pleural Mesothelioma. Front. Oncol. 2020, 10, 544789. [Google Scholar] [CrossRef]
- Lee, H.-S.; Jang, H.-J.; Choi, J.M.; Zhang, J.; de Rosen, V.L.; Wheeler, T.M.; Lee, J.-S.; Tu, T.; Jindra, P.T.; Kerman, R.H.; et al. Comprehensive Immunoproteogenomic Analyses of Malignant Pleural Mesothelioma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, G.J.; Rockwell, G.N.; Jensen, R.V.; Rheinwald, J.G.; Glickman, J.N.; Aronson, J.P.; Pottorf, B.J.; Nitz, M.D.; Richards, W.G.; Sugarbaker, D.J.; et al. Identification of Novel Candidate Oncogenes and Tumor Suppressors in Malignant Pleural Mesothelioma Using Large-Scale Transcriptional Profiling. Am. J. Pathol. 2005, 166, 1827–1840. [Google Scholar] [CrossRef]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A.; et al. Dissecting Heterogeneity in Malignant Pleural Mesothelioma through Histo-Molecular Gradients for Clinical Applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.S.; Righi, L.; Koeppen, H.; Zou, W.; Izzo, S.; Grosso, F.; Libener, R.; Loiacono, M.; Monica, V.; Buttigliero, C.; et al. Molecular and Histopathological Characterization of the Tumor Immune Microenvironment in Advanced Stage of Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qian, K.; Lu, G.; Chen, P.; Zhang, Y. Identification of Genes and Pathways Involved in Malignant Pleural Mesothelioma Using Bioinformatics Methods. BMC Med. Genom. 2021, 14, 104. [Google Scholar] [CrossRef]
- Duan, W.; Wang, K.; Duan, Y.; Chen, X.; Chu, X.; Hu, P.; Xiong, B. Combined Analysis of RNA Sequence and Microarray Data Reveals a Competing Endogenous RNA Network as Novel Prognostic Markers in Malignant Pleural Mesothelioma. Front. Oncol. 2021, 11, 615234. [Google Scholar] [CrossRef] [PubMed]
- Morani, F.; Bisceglia, L.; Rosini, G.; Mutti, L.; Melaiu, O.; Landi, S.; Gemignani, F. Identification of Overexpressed Genes in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2021, 22, 2738. [Google Scholar] [CrossRef]
- Liu, Z.; Klominek, J. Regulation of Matrix Metalloprotease Activity in Malignant Mesothelioma Cell Lines by Growth Factors. Thorax 2003, 58, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, S.; Giuffrida, A.; Fazi, M.; Coletti, A.; Palumbo, C.; Pass, H.I.; Procopio, A.; Modesti, A. Migration of Mesothelioma Cells Correlates with Histotype-Specific Synthesis of Extracellular Matrix. Int. J. Mol. Med. 1999, 4, 67–71. [Google Scholar] [CrossRef]
- Jagirdar, R.M.; Papazoglou, E.D.; Pitaraki, E.; Kouliou, O.A.; Rouka, E.; Giannakou, L.; Giannopoulos, S.; Sinis, S.I.; Hatzoglou, C.; Gourgoulianis, K.I.; et al. Cell and Extracellular Matrix Interaction Models in Benign Mesothelial and Malignant Pleural Mesothelioma Cells in 2D and 3D In-Vitro. Clin. Exp. Pharmacol. Physiol. 2021, 48, 543–552. [Google Scholar] [CrossRef]
- Abayasiriwardana, K.S.; Wood, M.K.; Prêle, C.M.; Birnie, K.A.; Robinson, B.W.; Laurent, G.J.; McAnulty, R.J.; Mutsaers, S.E. Inhibition of Collagen Production Delays Malignant Mesothelioma Tumor Growth in a Murine Model. Biochem. Biophys. Res. Commun. 2019, 510, 198–204. [Google Scholar] [CrossRef]
- Balancin, M.L.; Teodoro, W.R.; Farhat, C.; de Miranda, T.J.; Assato, A.K.; de Souza Silva, N.A.; Velosa, A.P.; Falzoni, R.; Ab’Saber, A.M.; Roden, A.C.; et al. An Integrative Histopathologic Clustering Model Based on Immuno-Matrix Elements to Predict the Risk of Death in Malignant Mesothelioma. Cancer Med. 2020, 9, 4836–4849. [Google Scholar] [CrossRef]
- Balancin, M.L.; Teodoro, W.R.; Baldavira, C.M.; Prieto, T.G.; Farhat, C.; Velosa, A.P.; da Costa Souza, P.; Yaegashi, L.B.; Ab’Saber, A.M.; Takagaki, T.Y.; et al. Different Histological Patterns of Type-V Collagen Levels Confer a Matrices-Privileged Tissue Microenvironment for Invasion in Malignant Tumors with Prognostic Value. Pathol. Res. Pract. 2020, 216, 153277. [Google Scholar] [CrossRef] [PubMed]
- Galateau Salle, F.; Le Stang, N.; Nicholson, A.G.; Pissaloux, D.; Churg, A.; Klebe, S.; Roggli, V.L.; Tazelaar, H.D.; Vignaud, J.M.; Attanoos, R.; et al. New Insights on Diagnostic Reproducibility of Biphasic Mesotheliomas: A Multi-Institutional Evaluation by the International Mesothelioma Panel From the MESOPATH Reference Center. J. Thorac. Oncol. 2018, 13, 1189–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galateau Salle, F.; Le Stang, N.; Tirode, F.; Courtiol, P.; Nicholson, A.G.; Tsao, M.-S.; Tazelaar, H.D.; Churg, A.; Dacic, S.; Roggli, V.; et al. Comprehensive Molecular and Pathologic Evaluation of Transitional Mesothelioma Assisted by Deep Learning Approach: A Multi-Institutional Study of the International Mesothelioma Panel from the MESOPATH Reference Center. J. Thorac. Oncol. 2020, 15, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A Peek into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis. Models Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, Y.; Enomoto, A.; Tsuyuki, Y.; Sato, K.; Iida, T.; Kobayashi, H.; Mizutani, Y.; Miyai, Y.; Hara, A.; Mii, S.; et al. Connective Tissue Growth Factor Produced by Cancer-Associated Fibroblasts Correlates with Poor Prognosis in Epithelioid Malignant Pleural Mesothelioma. Oncol. Rep. 2020, 44, 838–848. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF Receptors: Cancer Biology and Therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Marek, L.A.; Hinz, T.K.; von Mässenhausen, A.; Olszewski, K.A.; Kleczko, E.K.; Boehm, D.; Weiser-Evans, M.C.; Nemenoff, R.A.; Hoffmann, H.; Warth, A.; et al. Nonamplified FGFR1 Is a Growth Driver in Malignant Pleural Mesothelioma. Mol. Cancer Res. 2014, 12, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, C.; Sherk, C.; Fricko, M.; Ganji, G.; Barnette, M.; Hoang, B.; Tunstead, J.; Skedzielewski, T.; Alsaid, H.; Jucker, B.M.; et al. Inhibition of FGF/FGFR Autocrine Signaling in Mesothelioma with the FGF Ligand Trap, FP-1039/GSK3052230. Oncotarget 2016, 7, 39861–39871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.-S.; Creaney, J.; Chen, F.K.; Chin, W.L.; Muruganandan, S.; Arunachalam, S.; Attia, M.S.; Read, C.; Murray, K.; Millward, M.; et al. A Phase II Trial of Single Oral FGF Inhibitor, AZD4547, as Second or Third Line Therapy in Malignant Pleural Mesothelioma. Lung Cancer 2020, 140, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Hegmans, J.P.J.J.; Hemmes, A.; Hammad, H.; Boon, L.; Hoogsteden, H.C.; Lambrecht, B.N. Mesothelioma Environment Comprises Cytokines and T-Regulatory Cells That Suppress Immune Responses. Eur. Respir. J. 2006, 27, 1086–1095. [Google Scholar] [CrossRef]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Münzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast Growth Factor Receptor Inhibition Is Active against Mesothelioma and Synergizes with Radio- and Chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.; Vermeulen, P.B.; Van Marck, E. Angiogenic Cytokines in Mesothelioma: A Study of VEGF, FGF-1 and -2, and TGF Beta Expression. J. Pathol. 1999, 189, 72–78. [Google Scholar] [CrossRef]
- Righi, L.; Cavallo, M.C.; Gatti, G.; Monica, V.; Rapa, I.; Busso, S.; Albera, C.; Volante, M.; Scagliotti, G.V.; Papotti, M. Tumor/Stromal Caveolin-1 Expression Patterns in Pleural Mesothelioma Define a Subgroup of the Epithelial Histotype with Poorer Prognosis. Am. J. Clin. Pathol. 2014, 141, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Lolo, F.N.; Jiménez-Jiménez, V.; Sánchez-Álvarez, M.; Del Pozo, M.Á. Tumor-Stroma Biomechanical Crosstalk: A Perspective on the Role of Caveolin-1 in Tumor Progression. Cancer Metastasis Rev. 2020, 39, 485–503. [Google Scholar] [CrossRef]
- Albacete-Albacete, L.; Navarro-Lérida, I.; López, J.A.; Martín-Padura, I.; Astudillo, A.M.; Ferrarini, A.; Van-Der-Heyden, M.; Balsinde, J.; Orend, G.; Vázquez, J.; et al. ECM Deposition Is Driven by Caveolin-1-Dependent Regulation of Exosomal Biogenesis and Cargo Sorting. J. Cell Biol. 2020, 219, e202006178. [Google Scholar] [CrossRef]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-β Synergizes with Defects in the Hippo Pathway to Stimulate Human Malignant Mesothelioma Growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Yamashita, Y.; Chew, S.-H.; Akatsuka, S.; Ukai, S.; Wang, S.; Nagai, H.; Okazaki, Y.; Takahashi, T.; Toyokuni, S. Connective Tissue Growth Factor and β-Catenin Constitute an Autocrine Loop for Activation in Rat Sarcomatoid Mesothelioma. J. Pathol. 2014, 233, 402–414. [Google Scholar] [CrossRef]
- Ohara, Y.; Chew, S.H.; Misawa, N.; Wang, S.; Somiya, D.; Nakamura, K.; Kajiyama, H.; Kikkawa, F.; Tsuyuki, Y.; Jiang, L.; et al. Connective Tissue Growth Factor-Specific Monoclonal Antibody Inhibits Growth of Malignant Mesothelioma in an Orthotopic Mouse Model. Oncotarget 2018, 9, 18494–18509. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Fernández Pérez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an Anti-Connective Tissue Growth Factor Therapy, for Idiopathic Pulmonary Fibrosis (PRAISE): A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef]
- Picozzi, V.J.; Pipas, J.M.; Koong, A.; Giaccia, A.; Bahary, N.; Krishnamurthi, S.S.; Lopez, C.D.; O’Dwyer, P.J.; Modelska, K.; Poolman, V.; et al. FG-3019, a Human Monoclonal Antibody to CTGF, with Gemcitabine/Erlotinib in Patients with Locally Advanced or Metastatic Pancreatic Ductal Adenocarcinoma. JCO 2013, 31, 213. [Google Scholar] [CrossRef]
- Finger, E.C.; Cheng, C.-F.; Williams, T.R.; Rankin, E.B.; Bedogni, B.; Tachiki, L.; Spong, S.; Giaccia, A.J.; Powell, M.B. CTGF Is a Therapeutic Target for Metastatic Melanoma. Oncogene 2014, 33, 1093–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF Antagonism with MAb FG-3019 Enhances Chemotherapy Response without Increasing Drug Delivery in Murine Ductal Pancreas Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.M.; Bortolotto, C.; Benvenuti, S.; Lancia, A.; Filippi, A.R.; Stella, G.M. Malignant Pleural Mesothelioma: Genetic and Microenviromental Heterogeneity as an Unexpected Reading Frame and Therapeutic Challenge. Cancers 2020, 12, 1186. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Yang, H. Molecular Pathways: Targeting Mechanisms of Asbestos and Erionite Carcinogenesis in Mesothelioma. Clin. Cancer Res. 2012, 18, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Sayan, M.; Mossman, B.T. The NLRP3 Inflammasome in Pathogenic Particle and Fibre-Associated Lung Inflammation and Diseases. Part. Fibre Toxicol. 2016, 13, 51. [Google Scholar] [CrossRef] [Green Version]
- Kadariya, Y.; Menges, C.W.; Talarchek, J.; Cai, K.Q.; Klein-Szanto, A.J.; Pietrofesa, R.A.; Christofidou-Solomidou, M.; Cheung, M.; Mossman, B.T.; Shukla, A.; et al. Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev. Res. 2016, 9, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, R.F.; Iyer, L.L.; Holian, A. Asbestos Induces Apoptosis in Human Alveolar Macrophages. Am. J. Physiol. 1996, 271, L813–L819. [Google Scholar] [CrossRef]
- Jube, S.; Rivera, Z.S.; Bianchi, M.E.; Powers, A.; Wang, E.; Pagano, I.; Pass, H.I.; Gaudino, G.; Carbone, M.; Yang, H. Cancer Cell Secretion of the DAMP Protein HMGB1 Supports Progression in Malignant Mesothelioma. Cancer Res. 2012, 72, 3290–3301. [Google Scholar] [CrossRef] [Green Version]
- Tabata, C.; Shibata, E.; Tabata, R.; Kanemura, S.; Mikami, K.; Nogi, Y.; Masachika, E.; Nishizaki, T.; Nakano, T. Serum HMGB1 as a Prognostic Marker for Malignant Pleural Mesothelioma. BMC Cancer 2013, 13, 205. [Google Scholar] [CrossRef] [Green Version]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like Receptor 4 and High-Mobility Group Box-1 Are Involved in Ictogenesis and Can Be Targeted to Reduce Seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horio, D.; Minami, T.; Kitai, H.; Ishigaki, H.; Higashiguchi, Y.; Kondo, N.; Hirota, S.; Kitajima, K.; Nakajima, Y.; Koda, Y.; et al. Tumor-Associated Macrophage-Derived Inflammatory Cytokine Enhances Malignant Potential of Malignant Pleural Mesothelioma. Cancer Sci. 2020, 111, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Marcq, E.; Siozopoulou, V.; De Waele, J.; van Audenaerde, J.; Zwaenepoel, K.; Santermans, E.; Hens, N.; Pauwels, P.; van Meerbeeck, J.P.; Smits, E.L.J. Prognostic and Predictive Aspects of the Tumor Immune Microenvironment and Immune Checkpoints in Malignant Pleural Mesothelioma. Oncoimmunology 2017, 6, e1261241. [Google Scholar] [CrossRef] [Green Version]
- Burt, B.M.; Rodig, S.J.; Tilleman, T.R.; Elbardissi, A.W.; Bueno, R.; Sugarbaker, D.J. Circulating and Tumor-Infiltrating Myeloid Cells Predict Survival in Human Pleural Mesothelioma. Cancer 2011, 117, 5234–5244. [Google Scholar] [CrossRef]
- Cornelissen, R.; Lievense, L.A.; Robertus, J.-L.; Hendriks, R.W.; Hoogsteden, H.C.; Hegmans, J.P.J.J.; Aerts, J.G.J.V. Intratumoral Macrophage Phenotype and CD8+ T Lymphocytes as Potential Tools to Predict Local Tumor Outgrowth at the Intervention Site in Malignant Pleural Mesothelioma. Lung Cancer 2015, 88, 332–337. [Google Scholar] [CrossRef]
- Chu, G.J.; van Zandwijk, N.; Rasko, J.E.J. The Immune Microenvironment in Mesothelioma: Mechanisms of Resistance to Immunotherapy. Front. Oncol. 2019, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front. Immunol. 2017, 8, 892. [Google Scholar] [CrossRef]
- Alay, A.; Cordero, D.; Hijazo-Pechero, S.; Aliagas, E.; Lopez-Doriga, A.; Marín, R.; Palmero, R.; Llatjós, R.; Escobar, I.; Ramos, R.; et al. Integrative Transcriptome Analysis of Malignant Pleural Mesothelioma Reveals a Clinically Relevant Immune-Based Classification. J. Immunother. Cancer 2021, 9, e001601. [Google Scholar] [CrossRef]
- DeLong, P.; Carroll, R.G.; Henry, A.C.; Tanaka, T.; Ahmad, S.; Leibowitz, M.S.; Sterman, D.H.; June, C.H.; Albelda, S.M.; Vonderheide, R.H. Regulatory T Cells and Cytokines in Malignant Pleural Effusions Secondary to Mesothelioma and Carcinoma. Cancer Biol. Ther. 2005, 4, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, B.; Salcedo, A.; Lin, X.; Walkiewicz, M.; Murone, C.; Ameratunga, M.; Asadi, K.; Deb, S.; Barnett, S.A.; Knight, S.; et al. The Immune Microenvironment, Genome-Wide Copy Number Aberrations, and Survival in Mesothelioma. J. Thorac. Oncol. 2017, 12, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, P.; Ekeke, C.N.; Russell, K.L.; Butler, S.C.; Wang, Y.; Luketich, J.D.; Soloff, A.C.; Dhupar, R.; Lotze, M.T. Making Cold Malignant Pleural Effusions Hot: Driving Novel Immunotherapies. OncoImmunology 2019, 8, e1554969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.; Oizumi, S.; Kikuchi, E.; Shinagawa, N.; Konishi-Sakakibara, J.; Ishimine, A.; Aoe, K.; Gemba, K.; Kishimoto, T.; Torigoe, T.; et al. CD8+ Tumor-Infiltrating Lymphocytes Predict Favorable Prognosis in Malignant Pleural Mesothelioma after Resection. Cancer Immunol. Immunother. 2010, 59, 1543–1549. [Google Scholar] [CrossRef]
- Chee, S.J.; Lopez, M.; Mellows, T.; Gankande, S.; Moutasim, K.A.; Harris, S.; Clarke, J.; Vijayanand, P.; Thomas, G.J.; Ottensmeier, C.H. Evaluating the Effect of Immune Cells on the Outcome of Patients with Mesothelioma. Br. J. Cancer 2017, 117, 1341–1348. [Google Scholar] [CrossRef]
- Anraku, M.; Cunningham, K.S.; Yun, Z.; Tsao, M.-S.; Zhang, L.; Keshavjee, S.; Johnston, M.R.; de Perrot, M. Impact of Tumor-Infiltrating T Cells on Survival in Patients with Malignant Pleural Mesothelioma. J. Thorac. Cardiovasc. Surg. 2008, 135, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Pasello, G.; Zago, G.; Lunardi, F.; Urso, L.; Kern, I.; Vlacic, G.; Grosso, F.; Mencoboni, M.; Ceresoli, G.L.; Schiavon, M.; et al. Malignant Pleural Mesothelioma Immune Microenvironment and Checkpoint Expression: Correlation with Clinical–Pathological Features and Intratumor Heterogeneity over Time. Ann. Oncol. 2018, 29, 1258–1265. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Kopecka, J.; Napoli, F.; Pradotto, M.; Maletta, F.; Costardi, L.; Gagliasso, M.; Milosevic, V.; Ananthanarayanan, P.; Bironzo, P.; et al. Potential Diagnostic and Prognostic Role of Microenvironment in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2019, 14, 1458–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, N.; Vaira, V.; Righi, I.; Sajjadi, E.; Venetis, K.; Lopez, G.; Cattaneo, M.; Castellani, M.; Rosso, L.; Nosotti, M.; et al. Characterization of the Immune Microenvironment in Malignant Pleural Mesothelioma Reveals Prognostic Subgroups of Patients. Lung Cancer 2020, 150, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Manns, M.P.; Korangy, F. Myeloid Derived Suppressor Cells in Human Diseases. Int. Immunopharmacol. 2011, 11, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Mandruzzato, S.; Solito, S.; Falisi, E.; Francescato, S.; Chiarion-Sileni, V.; Mocellin, S.; Zanon, A.; Rossi, C.R.; Nitti, D.; Bronte, V.; et al. IL4Ralpha+ Myeloid-Derived Suppressor Cell Expansion in Cancer Patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef] [Green Version]
- Burt, B.M.; Bader, A.; Winter, D.; Rodig, S.J.; Bueno, R.; Sugarbaker, D.J. Expression of Interleukin-4 Receptor Alpha in Human Pleural Mesothelioma Is Associated with Poor Survival and Promotion of Tumor Inflammation. Clin. Cancer Res. 2012, 18, 1568–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minnema-Luiting, J.; Vroman, H.; Aerts, J.; Cornelissen, R. Heterogeneity in Immune Cell Content in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1041. [Google Scholar] [CrossRef] [Green Version]
- Khanna, S.; Graef, S.; Mussai, F.; Thomas, A.; Wali, N.; Yenidunya, B.G.; Yuan, C.; Morrow, B.; Zhang, J.; Korangy, F.; et al. Tumor-Derived GM-CSF Promotes Granulocyte Immunosuppression in Mesothelioma Patients. Clin. Cancer Res. 2018, 24, 2859–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upham, J.W.; Xi, Y. Dendritic Cells in Human Lung Disease: Recent Advances. Chest 2017, 151, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Lambrecht, B.N.; Hammad, H. Division of Labor between Lung Dendritic Cells and Macrophages in the Defense against Pulmonary Infections. Mucosal Immunol. 2013, 6, 464–473. [Google Scholar] [CrossRef]
- Kopf, M.; Schneider, C.; Nobs, S.P. The Development and Function of Lung-Resident Macrophages and Dendritic Cells. Nat. Immunol. 2015, 16, 36–44. [Google Scholar] [CrossRef]
- Lynch, J.P.; Mazzone, S.B.; Rogers, M.J.; Arikkatt, J.J.; Loh, Z.; Pritchard, A.L.; Upham, J.W.; Phipps, S. The Plasmacytoid Dendritic Cell: At the Cross-Roads in Asthma. Eur. Respir. J. 2014, 43, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid Dendritic Cell in Immunity and Cancer. J. Neuroimmunol. 2018, 322, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.K.; Mamotte, C.D.S.; Patel, P.; Yeoh, T.L.; Jackaman, C.; Nelson, D.J. Mesothelioma Tumor Cells Modulate Dendritic Cell Lipid Content, Phenotype and Function. PLoS ONE 2015, 10, e0123563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackaman, C.; Cornwall, S.; Graham, P.T.; Nelson, D.J. CD40-Activated B Cells Contribute to Mesothelioma Tumor Regression. Immunol. Cell Biol. 2011, 89, 255–267. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.I.; Ghafoor, A.; Sengupta, M.; Hassan, R. Malignant Mesothelioma: Advances in Immune Checkpoint Inhibitor and Mesothelin-Targeted Therapies. Cancer 2021, 127, 1010–1020. [Google Scholar] [CrossRef]
- Paver, E.C.; Cooper, W.A.; Colebatch, A.J.; Ferguson, P.M.; Hill, S.K.; Lum, T.; Shin, J.-S.; O’Toole, S.; Anderson, L.; Scolyer, R.A.; et al. Programmed Death Ligand-1 (PD-L1) as a Predictive Marker for Immunotherapy in Solid Tumours: A Guide to Immunohistochemistry Implementation and Interpretation. Pathology 2021, 53, 141–156. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune Checkpoint Inhibitors of PD-L1 as Cancer Therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- McCambridge, A.J.; Napolitano, A.; Mansfield, A.S.; Fennell, D.A.; Sekido, Y.; Nowak, A.K.; Reungwetwattana, T.; Mao, W.; Pass, H.I.; Carbone, M.; et al. Progress in the Management of Malignant Pleural Mesothelioma in 2017. J. Thorac. Oncol. 2018, 13, 606–623. [Google Scholar] [CrossRef] [Green Version]
- Scherpereel, A.; Wallyn, F.; Albelda, S.M.; Munck, C. Novel Therapies for Malignant Pleural Mesothelioma. Lancet Oncol. 2018, 19, e161–e172. [Google Scholar] [CrossRef]
- Jin, L.; Gu, W.; Li, X.; Xie, L.; Wang, L.; Chen, Z. PD-L1 and Prognosis in Patients with Malignant Pleural Mesothelioma: A Meta-Analysis and Bioinformatics Study. Adv. Med. Oncol. 2020, 12, 1758835920962362. [Google Scholar] [CrossRef]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 2017, 31, 501–515.e8. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, E.; Kajino, K.; Abe, M.; Ohtsuji, N.; Saeki, H.; Hlaing, M.T.; Hino, O. Expression Status of PD-L1 and B7-H3 in Mesothelioma. Pathol. Int. 2020, 70, 999–1008. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Dericks, L.; Schmid, R.A. CD44 and Its Ligand Hyaluronan as Potential Biomarkers in Malignant Pleural Mesothelioma: Evidence and Perspectives. Respir. Res. 2017, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Shingyoku, S.; Miyake, S.; Tanaka, A.; Fudesaka, S.; Shimizu, Y.; Yoshifuji, A.; Yamawaki, Y.; Yoshida, S.; Tanaka, S.; et al. Differential Regulation of the Sphere Formation and Maintenance of Cancer-Initiating Cells of Malignant Mesothelioma via CD44 and ALK4 Signaling Pathways. Oncogene 2018, 37, 6357–6367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Victoria Lai, W.; Adusumilli, P.S.; Desmeules, P.; Frosina, D.; Jungbluth, A.; Ni, A.; Eguchi, T.; Travis, W.D.; Ladanyi, M.; et al. V-Domain Ig-Containing Suppressor of T-Cell Activation (VISTA), a Potentially Targetable Immune Checkpoint Molecule, Is Highly Expressed in Epithelioid Malignant Pleural Mesothelioma. Mod. Pathol. 2020, 33, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcala, N.; Mangiante, L.; Le-Stang, N.; Gustafson, C.E.; Boyault, S.; Damiola, F.; Alcala, K.; Brevet, M.; Thivolet-Bejui, F.; Blanc-Fournier, C.; et al. Redefining Malignant Pleural Mesothelioma Types as a Continuum Uncovers Immune-Vascular Interactions. EBioMedicine 2019, 48, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Sekido, Y. Molecular Pathogenesis of Malignant Mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Strizzi, L.; Catalano, A.; Vianale, G.; Orecchia, S.; Casalini, A.; Tassi, G.; Puntoni, R.; Mutti, L.; Procopio, A. Vascular Endothelial Growth Factor Is an Autocrine Growth Factor in Human Malignant Mesothelioma. J. Pathol. 2001, 193, 468–475. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. VEGF-Targeted Therapy: Mechanisms of Anti-Tumour Activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- König, J.-E.; Tolnay, E.; Wiethege, T.; Müller, K.-M. Expression of Vascular Endothelial Growth Factor in Diffuse Malignant Pleural Mesothelioma. Virchows Arch. 1999, 435, 8–12. [Google Scholar] [CrossRef]
- Ohta, Y.; Shridhar, V.; Bright, R.K.; Kalemkerian, G.P.; Du, W.; Carbone, M.; Watanabe, Y.; Pass, H.I. VEGF and VEGF Type C Play an Important Role in Angiogenesis and Lymphangiogenesis in Human Malignant Mesothelioma Tumours. Br. J. Cancer 1999, 81, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Linder, C.; Linder, S.; Munck-Wikland, E.; Strander, H. Independent Expression of Serum Vascular Endothelial Growth Factor (VEGF) and Basic Fibroblast Growth Factor (BFGF) in Patients with Carcinoma and Sarcoma. Anticancer Res. 1998, 18, 2063–2068. [Google Scholar] [PubMed]
- Antony, V.B.; Hott, J.W.; Godbey, S.W.; Holm, K. Angiogenesis in Mesotheliomas. Role of Mesothelial Cell Derived IL-8. Chest 1996, 109, 21S–22S. [Google Scholar] [CrossRef]
- Nowak, A.K.; Brosseau, S.; Cook, A.; Zalcman, G. Antiangiogeneic Strategies in Mesothelioma. Front. Oncol. 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- De Marinis, F.; Bria, E.; Ciardiello, F.; Crinò, L.; Douillard, J.Y.; Griesinger, F.; Lambrechts, D.; Perol, M.; Ramalingam, S.S.; Smit, E.F.; et al. International Experts Panel Meeting of the Italian Association of Thoracic Oncology on Antiangiogenetic Drugs for Non-Small Cell Lung Cancer: Realities and Hopes. J. Thorac. Oncol. 2016, 11, 1153–1169. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.; Grosso, F.; Steele, N.; Novello, S.; Popat, S.; Greillier, L.; John, T.; Leighl, N.; Reck, M.; Pavlakis, N.; et al. MA 19.03 Nintedanib + Pemetrexed/Cisplatin in Malignant Pleural Mesothelioma (MPM): Phase II Biomarker Data from the LUME-Meso Study. J. Thorac. Oncol. 2017, 12, S1884. [Google Scholar] [CrossRef]
- Chia, P.L.; Russell, P.; Asadi, K.; Thapa, B.; Gebski, V.; Murone, C.; Walkiewicz, M.; Eriksson, U.; Scott, A.M.; John, T. Analysis of Angiogenic and Stromal Biomarkers in a Large Malignant Mesothelioma Cohort. Lung Cancer 2020, 150, 1–8. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napoli, F.; Listì, A.; Zambelli, V.; Witel, G.; Bironzo, P.; Papotti, M.; Volante, M.; Scagliotti, G.; Righi, L. Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma. Cancers 2021, 13, 2564. https://doi.org/10.3390/cancers13112564
Napoli F, Listì A, Zambelli V, Witel G, Bironzo P, Papotti M, Volante M, Scagliotti G, Righi L. Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma. Cancers. 2021; 13(11):2564. https://doi.org/10.3390/cancers13112564
Chicago/Turabian StyleNapoli, Francesca, Angela Listì, Vanessa Zambelli, Gianluca Witel, Paolo Bironzo, Mauro Papotti, Marco Volante, Giorgio Scagliotti, and Luisella Righi. 2021. "Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma" Cancers 13, no. 11: 2564. https://doi.org/10.3390/cancers13112564
APA StyleNapoli, F., Listì, A., Zambelli, V., Witel, G., Bironzo, P., Papotti, M., Volante, M., Scagliotti, G., & Righi, L. (2021). Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma. Cancers, 13(11), 2564. https://doi.org/10.3390/cancers13112564