
Carcinogenesis of Triple-Negative Breast Cancer and Sex Steroid Hormones


Abstract
:Simple Summary
Abstract
1. Introduction
2. Subclassification of TNBC
3. Sex Steroid Hormones
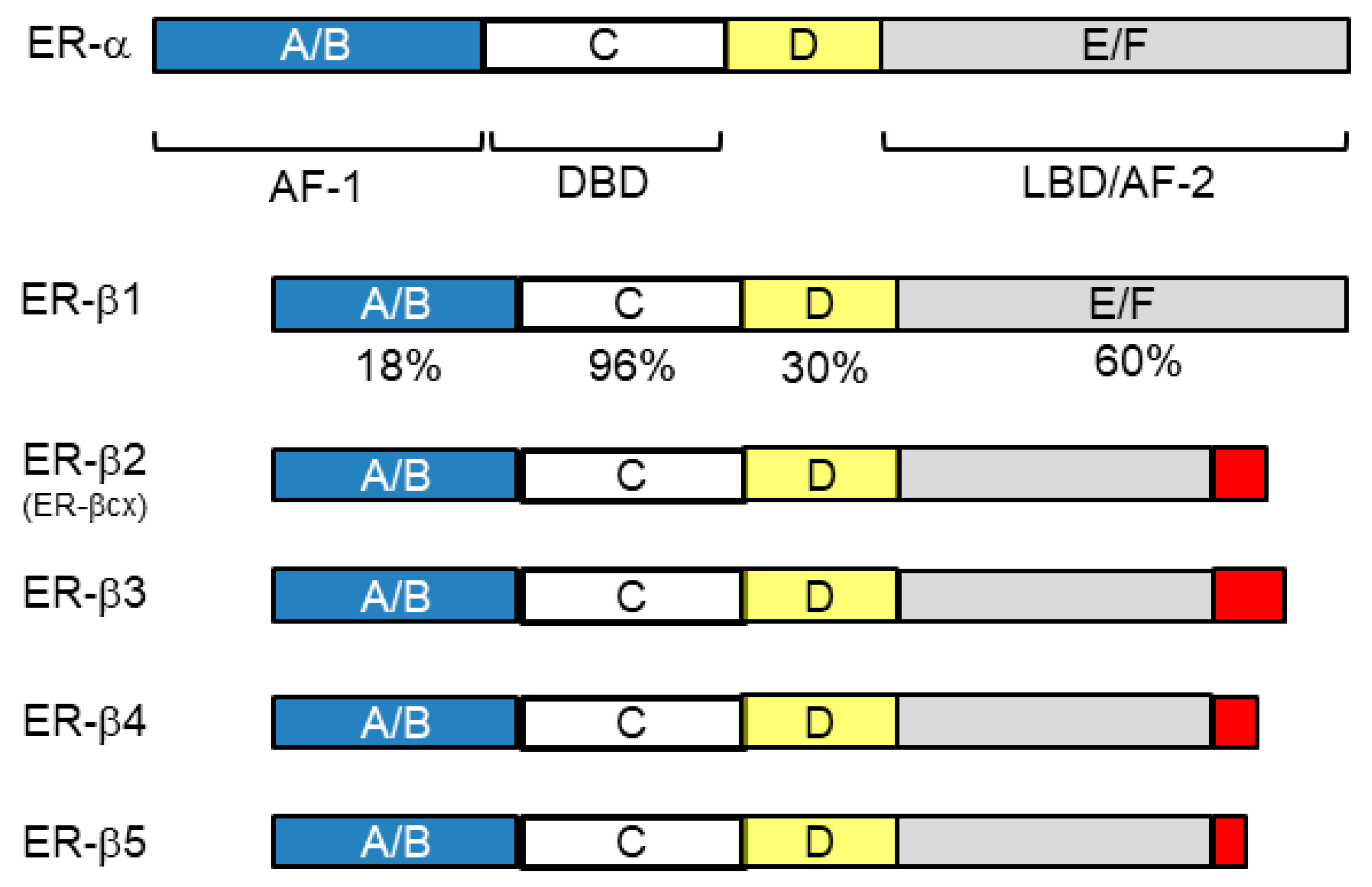
4. Sex Steroid Hormone Receptors Other Than ER-α and PgR
4.1. Nuclear Receptors (ER-β and AR)
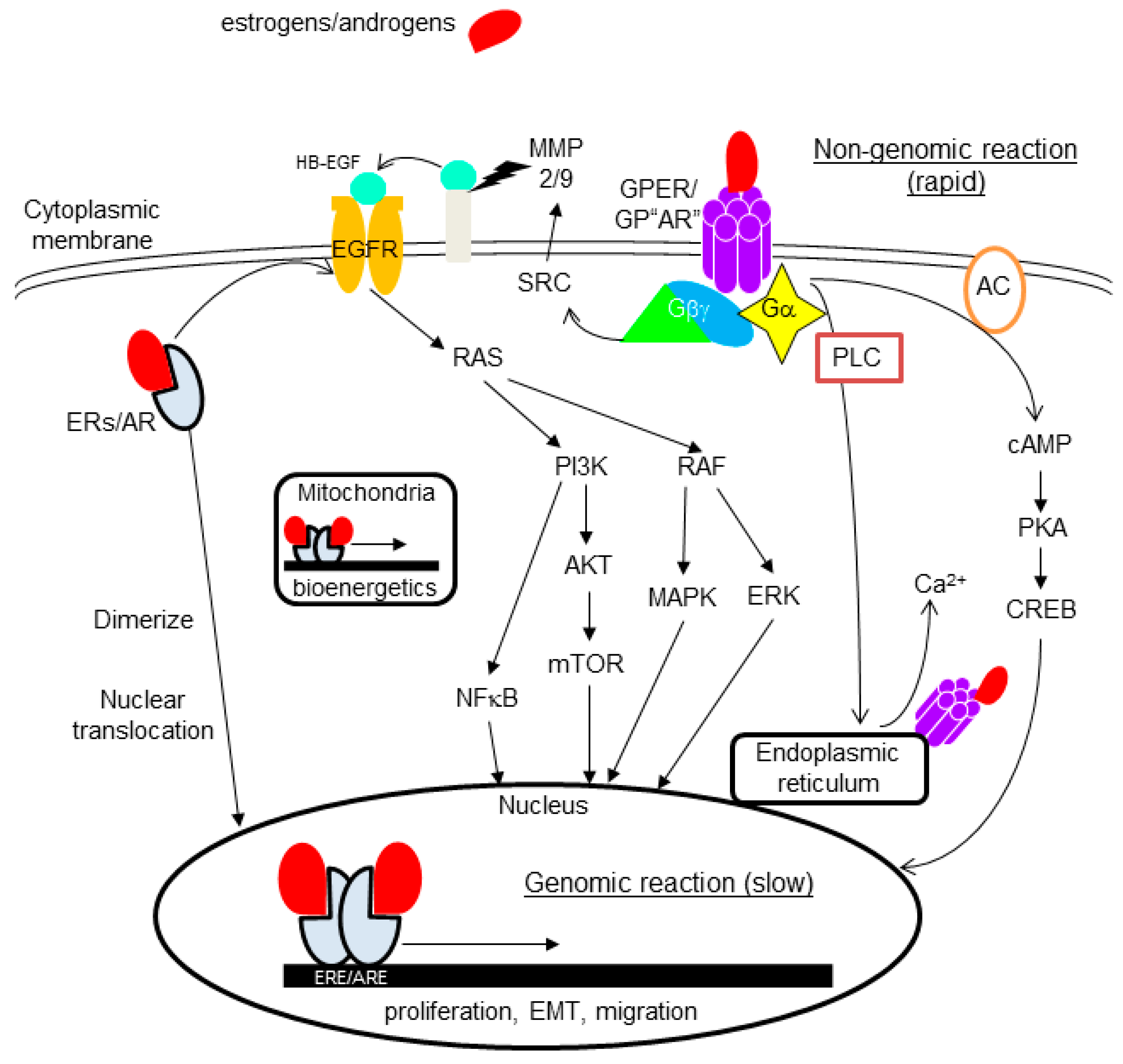
4.2. Non-Nuclear-Located Sex Steroid Hormone Receptors
4.2.1. Membrane-Bound Receptors
4.2.2. Cytoplasmic Receptors
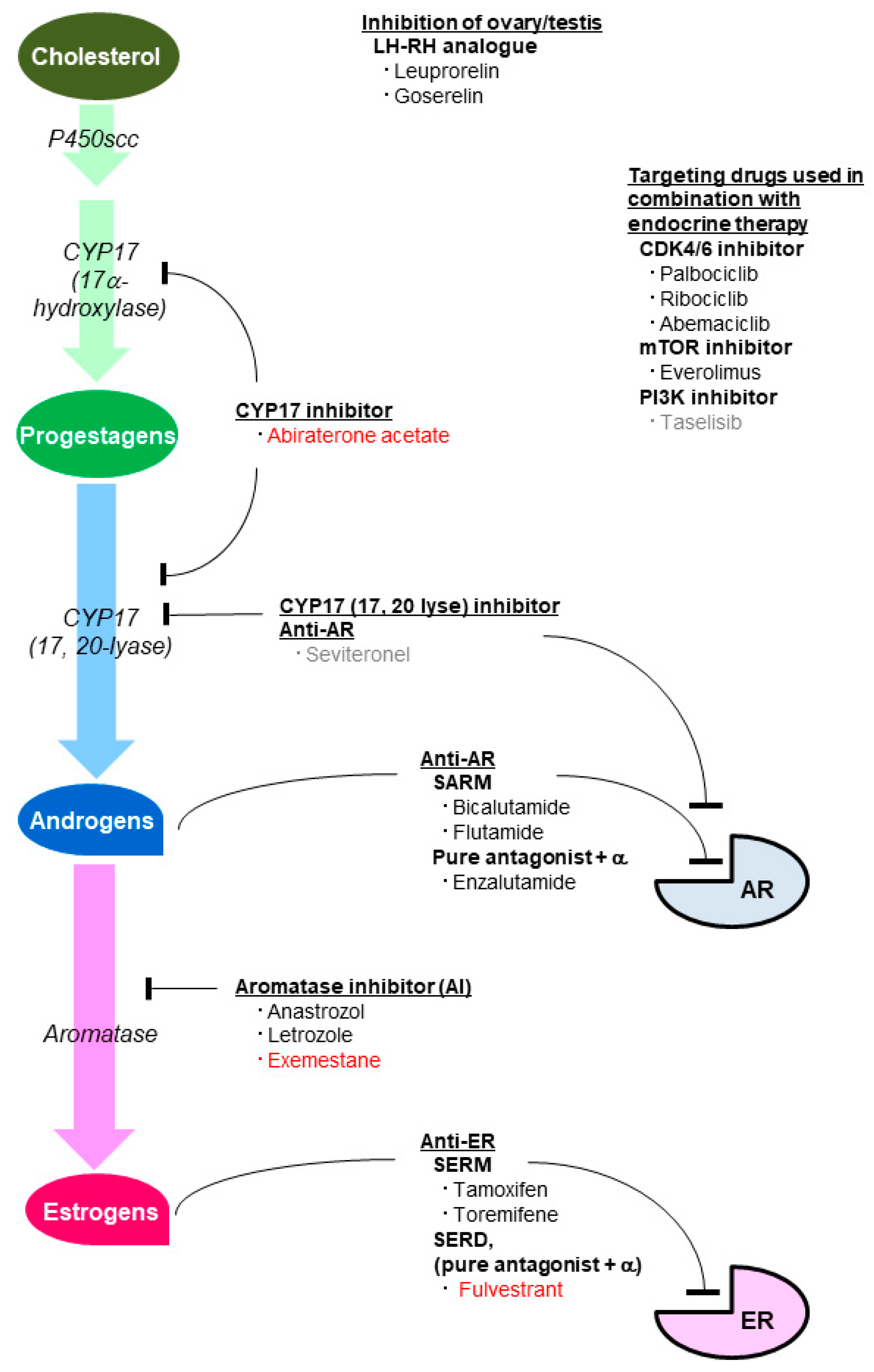
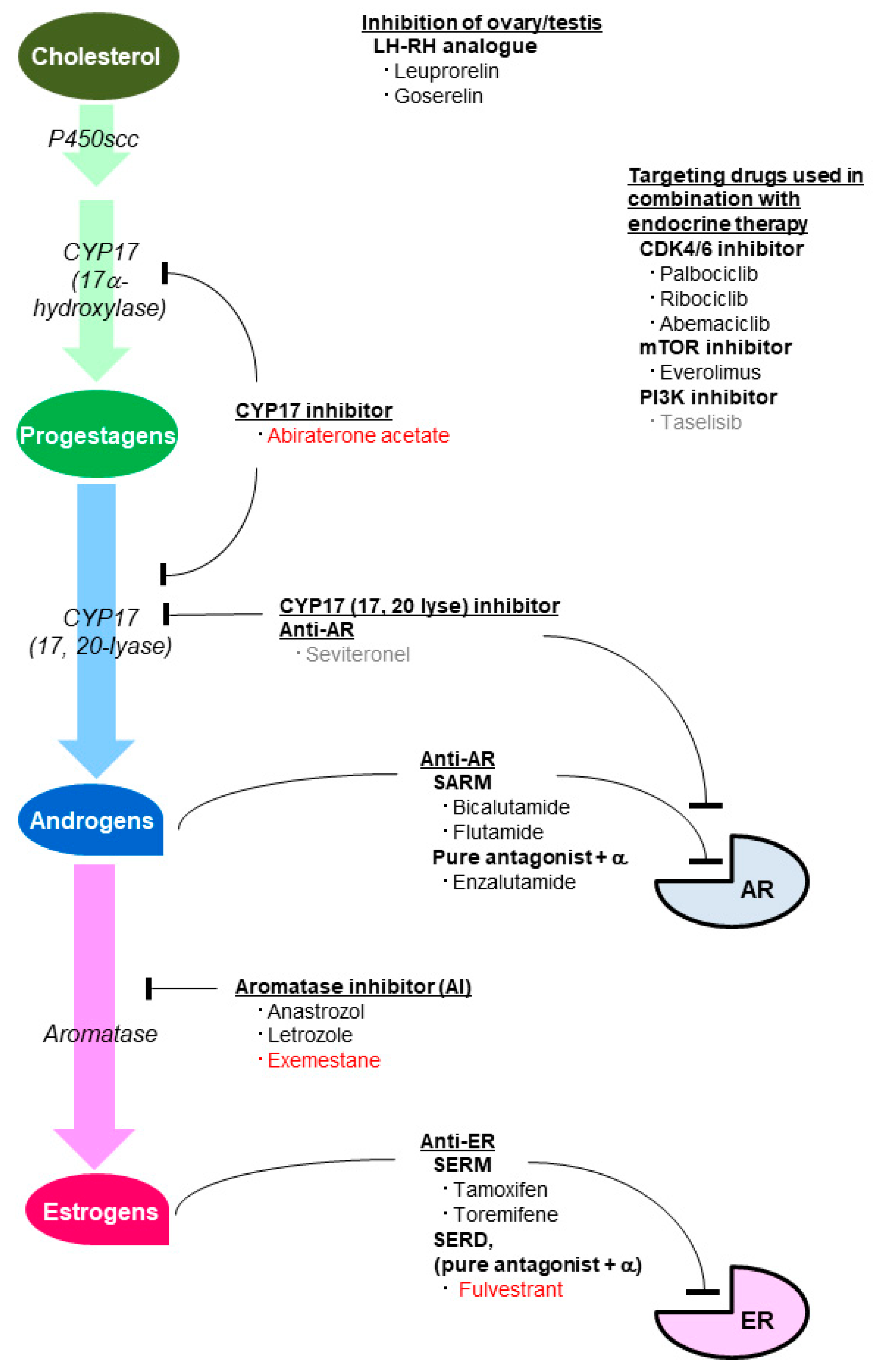
5. Agents Inhibiting the Effect of Sex Steroid Hormones
5.1. Agents Inhibiting the Estrogenic Effect
5.2. Agents Inhibiting the Androgenic Effect
6. The Role of Sex Steroid Hormones in TNBC in a Preclinical Setting
6.1. The Role of AR in TNBC
6.2. The Role of ER-β in TNBC
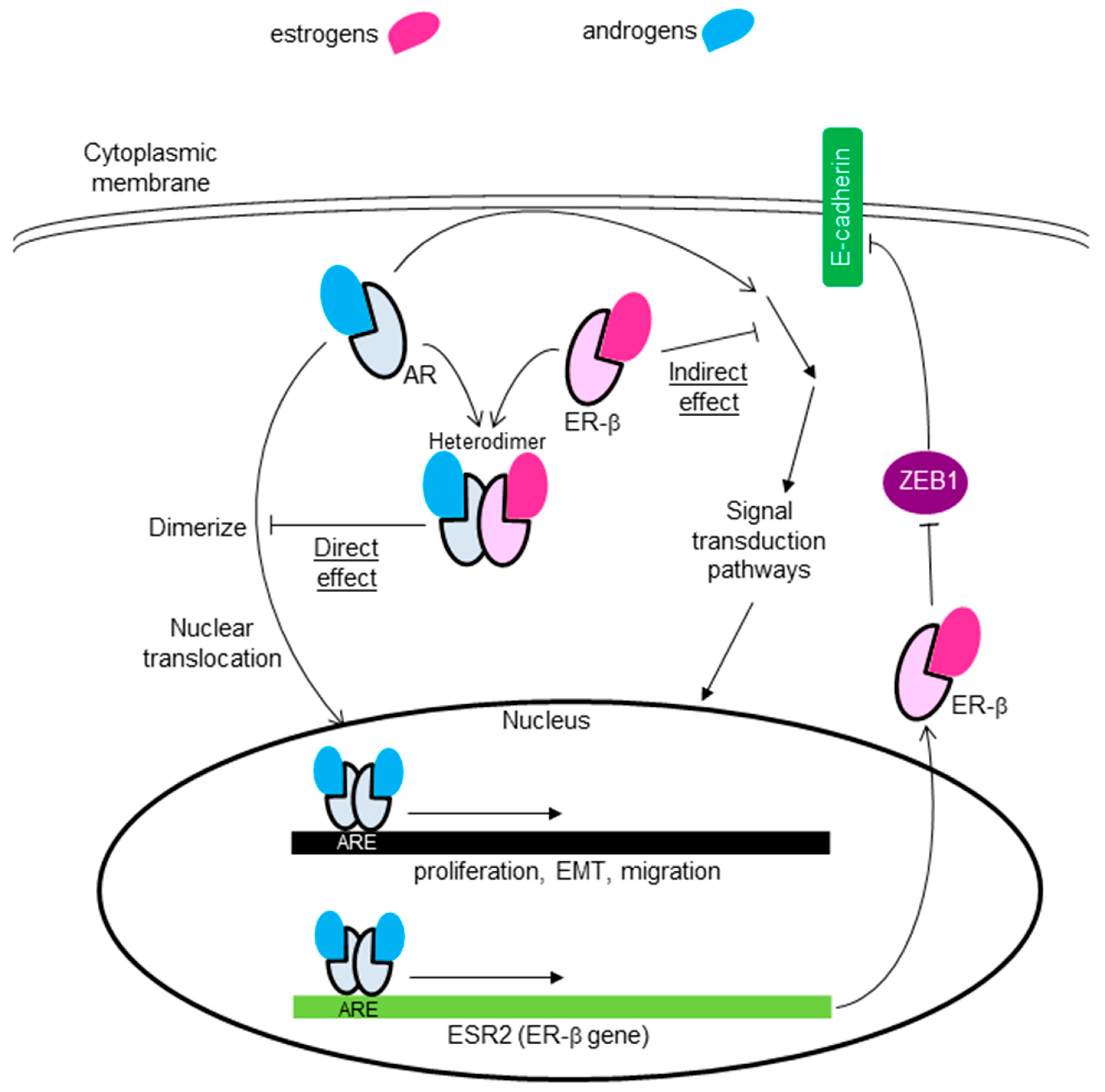
6.3. Crosstalk between AR and ER-β
6.4. The Role of Non-Nuclear-Located Receptors in TNBC
7. The Role of Each Sex Steroid Hormone in TNBC in a Clinical Setting
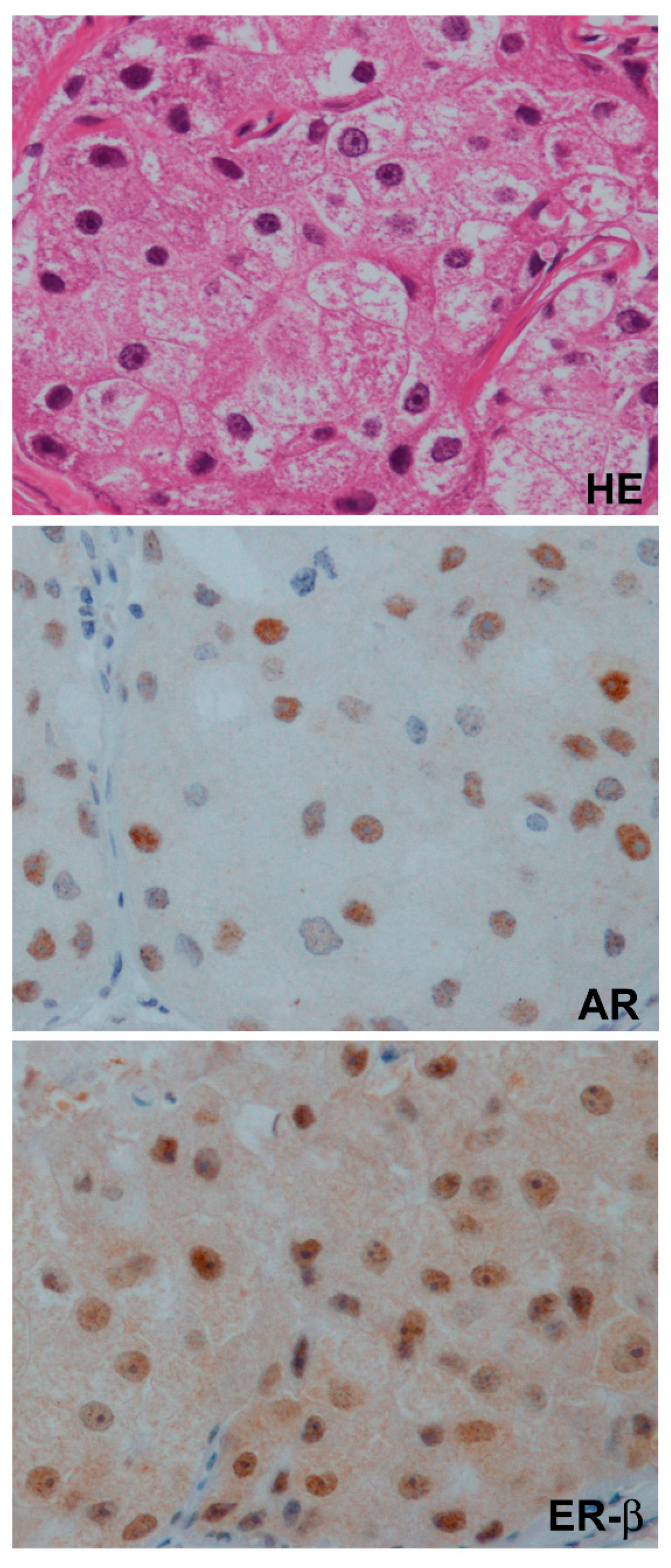
7.1. The Role of AR in a Clinical Setting
7.2. The Role of ER-β in a Clinical Setting
7.3. Correlation of AR and ER-β in Clinical TNBC
7.4. The Role of Non-Nuclear Receptors in a Clinical Setting
7.5. Endocrine Therapy for Patients with TNBC
8. Materials and Methods
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burstein, H.J.; Curigliano, G.; Loibl, S.; Dubsky, P.; Gnant, M.; Poortmans, P.; Colleoni, M.; Denkert, C.; Piccart-Gebhart, M.; Regan, M.; et al. Estimating the Benefits of Therapy for Early-Stage Breast Cancer: The St. Gallen International Consensus Guidelines for the Primary Therapy of Early Breast Cancer 2019. Ann. Oncol. 2019, 30, 1541–1557. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease. Nat. Rev. Clin. Oncol. 2016, 13, 674. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; Von Minckwitz, G.; Tutt, A.G. Breast Cancer 3 Molecular Alterations in Triple-Negative Breast Cancer- the Road to New Treatment Strategies. Lancet 2017, 389, 2430. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Oakman, C.; Viale, G.; Di Leo, A. Management of Triple Negative Breast Cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.S.; Robertson, J.F.; Ellis, I.O. Prognostic Markers in Triple-Negative Breast Cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- WHO Classification of Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019.
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; De Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; De Santo, I.; Del Mastro, L.; et al. Androgen Receptor in Triple Negative Breast Cancer: A Potential Target for the Targetless Subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II Trial of Bicalutamide in Patients with Androgen Receptor-Positive, Estrogen Receptor-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A Phase II Trial of Abiraterone Acetate Plus Prednisone in Patients with Triple-Negative Androgen Receptor Positive Locally Advanced or Metastatic Breast Cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Ito, Y.; Akiyama, F. Clinical Importance of Androgen Receptor in Breast Cancer Patients Treated with Adjuvant Tamoxifen Monotherapy. Breast Cancer 2013, 20, 323–330. [Google Scholar] [CrossRef]
- Bozovic-Spasojevic, I.; Zardavas, D.; Brohee, S.; Ameye, L.; Fumagalli, D.; Ades, F.; de Azambuja, E.; Bareche, Y.; Piccart, M.; Paesmans, M.; et al. The Prognostic Role of Androgen Receptor in Patients with Early-Stage Breast Cancer: A Meta-Analysis of Clinical and Gene Expression Data. Clin. Cancer Res. 2017, 23, 2702–2712. [Google Scholar] [CrossRef] [Green Version]
- Astvatsaturyan, K.; Yue, Y.; Walts, A.E.; Bose, S. Androgen Receptor Positive Triple Negative Breast Cancer: Clinicopathologic, Prognostic, and Predictive Features. PLoS ONE 2018, 13, e0197827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.E.; Kang, S.H.; Lee, S.J.; Bae, Y.K. Androgen Receptor Expression Predicts Decreased Survival in Early Stage Triple-Negative Breast Cancer. Ann. Surg. Oncol. 2015, 22, 82–89. [Google Scholar] [CrossRef]
- Dieci, M.V.; Tsvetkova, V.; Griguolo, G.; Miglietta, F.; Mantiero, M.; Tasca, G.; Cumerlato, E.; Giorgi, C.A.; Giarratano, T.; Faggioni, G.; et al. Androgen Receptor Expression and Association with Distant Disease-Free Survival in Triple Negative Breast Cancer: Analysis of 263 Patients Treated with Standard Therapy for Stage I-III Disease. Front. Oncol. 2019, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Grellety, T. Androgen Receptor-Positive Triple Negative Breast Cancer: From Biology to Therapy. Bull. Cancer 2020. [Google Scholar] [CrossRef]
- Guiu, S.; Charon-Barra, C.; Vernerey, D.; Fumoleau, P.; Campone, M.; Spielmann, M.; Roche, H.; Mesleard, C.; Arnould, L.; Lemonnier, J.; et al. Coexpression of Androgen Receptor and FOXA1 in Nonmetastatic Triple-Negative Breast Cancer: Ancillary Study from PACS08 Trial. Future Oncol. 2015, 11, 2283–2297. [Google Scholar] [CrossRef] [PubMed]
- Kensler, K.H.; Poole, E.M.; Heng, Y.J.; Collins, L.C.; Glass, B.; Beck, A.H.; Hazra, A.; Rosner, B.A.; Eliassen, A.H.; Hankinson, S.E.; et al. Androgen Receptor Expression and Breast Cancer Survival: Results from the Nurses’ Health Studies. J. Natl. Cancer Inst. 2019, 111, 700–708. [Google Scholar] [CrossRef]
- Sutton, L.M.; Cao, D.; Sarode, V.; Molberg, K.H.; Torgbe, K.; Haley, B.; Peng, Y. Decreased Androgen Receptor Expression is Associated with Distant Metastases in Patients with Androgen Receptor-Expressing Triple-Negative Breast Carcinoma. Am. J. Clin. Pathol. 2012, 138, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Vera-Badillo, F.E.; Templeton, A.J.; de Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen Receptor Expression and Outcomes in Early Breast Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, djt319. [Google Scholar] [CrossRef] [PubMed]
- Anestis, A.; Karamouzis, M.V.; Dalagiorgou, G.; Papavassiliou, A.G. Is Androgen Receptor Targeting an Emerging Treatment Strategy for Triple Negative Breast Cancer? Cancer Treat. Rev. 2015, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Takubo, K.; Matsuura, M.; Ito, Y.; Akiyama, F.; Sakamoto, G. Clinical Importance of Estrogen Receptor-Beta Evaluation in Breast Cancer Patients Treated with Adjuvant Tamoxifen Therapy. J. Clin. Oncol. 2008, 26, 3727–3734. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, C.; Chen, K.; Tang, H.; Tang, J.; Song, C.; Xie, X. ERβ1 Inversely Correlates with PTEN/PI3K/AKT Pathway and Predicts a Favorable Prognosis in Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2015, 152, 255–269. [Google Scholar] [CrossRef]
- Sellitto, A.; D’Agostino, Y.; Alexandrova, E.; Lamberti, J.; Pecoraro, G.; Memoli, D.; Rocco, D.; Coviello, E.; Giurato, G.; Nassa, G.; et al. Insights into the Role of Estrogen Receptor Beta in Triple-Negative Breast Cancer. Cancers 2020, 12, 1477. [Google Scholar] [CrossRef]
- Shanle, E.K.; Onitilo, A.A.; Huang, W.; Kim, K.; Zang, C.; Engel, J.M.; Xu, W.; Wisinski, K.B. Prognostic Significance of Full-Length Estrogen Receptor Beta Expression in Stage I-III Triple Negative Breast Cancer. Am. J. Transl. Res. 2015, 7, 1246–1259. [Google Scholar]
- Guo, L.; Zhu, Q.; Aisimutuola, M.; Yilamu, D.; Liu, S.; Jakulin, A. Expression and Prognostic Value of Estrogen Receptor Β in Patients with Triple-Negative and Triple-Positive Breast Cancer. Exp. Ther. Med. 2015, 9, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Suman, V.J.; Subramaniam, M.; Wu, X.; Negron, V.; Gingery, A.; Pitel, K.S.; Shah, S.S.; Cunliffe, H.E.; McCullough, A.E.; et al. ERβ1: Characterization, Prognosis, and Evaluation of Treatment Strategies in ERα-Positive and -Negative Breast Cancer. BMC Cancer 2014, 14, 749. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Silva, C.D.; Villegas-Pineda, J.C.; Pereira-Suarez, A.L. Expression and Role of the G Protein-Coupled Estrogen Receptor (GPR30/GPER) in the Development and Immune Response in Female Reproductive Cancers. Front. Endocrinol. 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.H.; Chu, N.M.; Lin, Y.F.; Kao, S.H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiman, J.; Peralta, E.A.; Louis, S.; Kamel, O. Biology of the Estrogen Receptor, GPR30, in Triple Negative Breast Cancer. Am. J. Surg. 2013, 206, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Liu, M.; Luo, H.; Wu, C.; Tang, X.; Tang, S.; Hu, P.; Yan, Y.; Wang, Z.; Tu, G. GPER Mediates Enhanced Cell Viability and Motility Via Non-Genomic Signaling Induced by 17β-Estradiol in Triple-Negative Breast Cancer Cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 392–403. [Google Scholar] [CrossRef]
- Chen, Z.; Wei, W.; Jiang, G.; Liu, H.; Wei, W.; Yang, X.; Wu, Y.; Liu, H.; Wong, C.K.C.; Du, J.; et al. Activation of GPER Suppresses Epithelial Mesenchymal Transition of Triple Negative Breast Cancer Cells Via NF-κB Signals. Mol. Oncol. 2016, 10, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Chen, Z.; Jiang, G.; Zhou, Y.; Liu, Q.; Su, Q.; Wei, W.; Du, J.; Wang, H. Activation of GPER Suppresses Migration and Angiogenesis of Triple Negative Breast Cancer Via Inhibition of NF-κB/IL-6 Signals. Cancer Lett. 2017, 386, 12–23. [Google Scholar] [CrossRef]
- Hanukoglu, I. Steroidogenic Enzymes: Structure, Function, and Role in Regulation of Steroid Hormone Biosynthesis. J. Steroid Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.M.; Gauthier, S.; Labrie, F. Androgen Receptor Antagonists (Antiandrogens): Structure-Activity Relationships. Curr. Med. Chem. 2000, 7, 211–247. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.H.; Hales, D.B. Overview of Steroidogenic Enzymes in the Pathway from Cholesterol to Active Steroid Hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R.; Hawkins, R.A.; Forrest, A.P. Significance of Aromatase Activity in Human Breast Cancer. Cancer Res. 1982, 42, 3365s–3368s. [Google Scholar]
- Honma, N.; Saji, S.; Hirose, M.; Horiguchi, S.; Kuroi, K.; Hayashi, S.; Utsumi, T.; Harada, N. Sex Steroid Hormones in Pairs of Tumor and Serum from Breast Cancer Patients and Pathobiological Role of Androstene-3β, 17β-Diol. Cancer Sci. 2011, 102, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Honma, N.; Saji, S.; Mikami, T.; Yoshimura, N.; Mori, S.; Saito, Y.; Murayama, S.; Harada, N. Estrogen-Related Factors in the Frontal Lobe of Alzheimer’s Disease Patients and Importance of Body Mass Index. Sci. Rep. 2017, 7, 726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; He, P.; Cui, J.; Staufenbiel, M.; Harada, N.; Shen, Y. Brain Endogenous Estrogen Levels Determine Responses to Estrogen Replacement Therapy Via Regulation of BACE1 and NEP in Female Alzheimer’s Transgenic Mice. Mol. Neurobiol. 2013, 47, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Mahabir, S.; Baer, D.J.; Johnson, L.L.; Hartman, T.J.; Dorgan, J.F.; Campbell, W.S.; Clevidence, B.A.; Taylor, P.R. Usefulness of Body Mass Index as a Sufficient Adiposity Measurement for Sex Hormone Concentration Associations in Postmenopausal Women. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2502–2507. [Google Scholar] [CrossRef] [Green Version]
- Khosla, S.; Melton, L.J.; Atkinson, E.J.; O’Fallon, W.M. Relationship of Serum Sex Steroid Levels to Longitudinal Changes in Bone Density in Young Versus Elderly Men. J. Clin. Endocrinol. Metab. 2001, 86, 3555–3561. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the Treatment of Women with Early Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Eusebi, V.; Magalhaes, F.; Azzopardi, J.G. Pleomorphic Lobular Carcinoma of the Breast: An Aggressive Tumor Showing Apocrine Differentiation. Hum. Pathol. 1992, 23, 655–662. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes in a Phase III Randomized Adjuvant Breast Cancer Trial in Node-Positive Breast Cancer Comparing the Addition of Docetaxel to Doxorubicin with Doxorubicin-Based Chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Moon, B.I.; Lim, W.; Park, S.; Cho, M.S.; Sung, S.H. Feasibility of Classification of Triple Negative Breast Cancer by Immunohistochemical Surrogate Markers. Clin. Breast Cancer 2018, 18, e1123–e1132. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Patients with Early-Stage Triple-Negative Breast Cancers (TNBC) Who did Not Receive Adjuvant Chemotherapy. Ann. Oncol. 2019, 30, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Paruthiyil, S.; Cvoro, A.; Zhao, X.; Wu, Z.; Sui, Y.; Staub, R.E.; Baggett, S.; Herber, C.B.; Griffin, C.; Tagliaferri, M.; et al. Drug and Cell Type-Specific Regulation of Genes with Different Classes of Estrogen Receptor Beta-Selective Agonists. PLoS ONE 2009, 4, e6271. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Martínez-Archundia, M.; Maroun, R.C.; Ceballos-Reyes, G.; Fragoso-Vázquez, M.J.; González-Juárez, D.E.; Correa-Basurto, J. Deciphering the GPER/GPR30-Agonist and Antagonists Interactions using Molecular Modeling Studies, Molecular Dynamics, and Docking Simulations. J. Biomol. Struct. Dyn. 2015, 33, 2161–2172. [Google Scholar] [CrossRef]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G Protein-Coupled Estrogen Receptor GPER in Metabolic Regulation. J. Steroid Biochem. Mol. Biol. 2018, 176, 31–37. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, H.; He, J.; Wu, X.; Wang, L.; Liu, X.; Lin, H. Interaction of Nuclear ERs and GPER in Vitellogenesis in Zebrafish. J. Steroid Biochem. Mol. Biol. 2019, 189, 10–18. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y. Membrane Progesterone Receptors: Evidence for Neuroprotective, Neurosteroid Signaling and Neuroendocrine Functions in Neuronal Cells. Neuroendocrinology 2012, 96, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and Characterization of Human Estrogen Receptor Beta Isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef]
- Shah, P.D.; Gucalp, A.; Traina, T.A. The Role of the Androgen Receptor in Triple-Negative Breast Cancer. Womens Health 2013, 9, 351–360. [Google Scholar] [CrossRef]
- Hamilton, N.; Marquez-Garban, D.; Mah, V.H.; Elshimali, Y.; Elashoff, D.; Garon, E.B.; Vadgama, J.; Pietras, R. Estrogen Receptor-Beta and the Insulin-Like Growth Factor Axis as Potential Therapeutic Targets for Triple-Negative Breast Cancer. Crit. Rev. Oncog. 2015, 20, 373–390. [Google Scholar] [CrossRef] [Green Version]
- Haldosén, L.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor Beta in Breast Cancer. Mol. Cell Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. Mol. Biol. 2019, 191, 105312. [Google Scholar] [CrossRef]
- Katsu, Y.; Matsubara, K.; Kohno, S.; Matsuda, Y.; Toriba, M.; Oka, K.; Guillette, L.J.; Ohta, Y.; Iguchi, T. Molecular Cloning, Characterization, and Chromosome Mapping of Reptilian Estrogen Receptors. Endocrinology 2010, 151, 5710–5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Karthik, G.M.; Lovrot, J.; Haglund, F.; Rosin, G.; Katchy, A.; Zhang, X.; Viberg, L.; Frisell, J.; Williams, C.; et al. Estrogen Receptor Beta as a Therapeutic Target in Breast Cancer Stem Cells. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef]
- Kalyvianaki, K.; Panagiotopoulos, A.A.; Malamos, P.; Moustou, E.; Tzardi, M.; Stathopoulos, E.N.; Ioannidis, G.S.; Marias, K.; Notas, G.; Theodoropoulos, P.A.; et al. Membrane Androgen Receptors (OXER1, GPRC6A AND ZIP9) in Prostate and Breast Cancer: A Comparative Study of their Expression. Steroids 2019, 142, 100–108. [Google Scholar] [CrossRef]
- Rosenfeld, C.S.; Cooke, P.S. Endocrine Disruption through Membrane Estrogen Receptors and Novel Pathways Leading to Rapid Toxicological and Epigenetic Effects. J. Steroid Biochem. Mol. Biol. 2019, 187, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Honma, N.; Saji, S.; Kurabayashi, R.; Aida, J.; Arai, T.; Horii, R.; Akiyama, F.; Iwase, T.; Harada, N.; Younes, M.; et al. Oestrogen Receptor-Beta1 but Not Oestrogen Receptor-Betacx is of Prognostic Value in Apocrine Carcinoma of the Breast. APMIS 2008, 116, 923–930. [Google Scholar] [CrossRef]
- Honma, N.; Takubo, K.; Akiyama, F.; Kasumi, F.; Sawabe, M.; Arai, T.; Hosoi, T.; Yoshimura, N.; Harada, N.; Younes, M.; et al. Expression of Oestrogen Receptor-Beta in Apocrine Carcinomas of the Breast. Histopathology 2007, 50, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.; Tzeng, C.; Yu, C.; Wang, Y.; Kao, S. Estrogen Receptor-Β in Mitochondria: Implications for Mitochondrial Bioenergetics and Tumorigenesis. Ann. N. Y. Acad. Sci. 2015, 1350, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G Protein-Coupled Estrogen Receptor as a Modulator of Neoplastic Transformation. Mol. Cell Endocrinol. 2016, 429, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, A.; Ignatov, T.; Roessner, A.; Costa, S.D.; Kalinski, T. Role of GPR30 in the Mechanisms of Tamoxifen Resistance in Breast Cancer MCF-7 Cells. Breast Cancer Res. Treat. 2010, 123, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, A.; Ignatov, T.; Weissenborn, C.; Eggemann, H.; Bischoff, J.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T. G-Protein-Coupled Estrogen Receptor GPR30 and Tamoxifen Resistance in Breast Cancer. Breast Cancer Res. Treat. 2011, 128, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an Initiator of Tamoxifen Resistance in Hormone-Dependent Breast Cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.W.; Maggiolini, M.; et al. Tamoxifen through GPER Upregulates Aromatase Expression: A Novel Mechanism Sustaining Tamoxifen-Resistant Breast Cancer Cell Growth. Breast Cancer Res. Treat. 2014, 146, 273–285. [Google Scholar] [CrossRef]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Roberto, M.; Astone, A.; Botticelli, A.; Carbognin, L.; Cassano, A.; D’Auria, G.; Fabbri, A.; Fabi, A.; Gamucci, T.; Krasniqi, E.; et al. CDK4/6 Inhibitor Treatments in Patients with Hormone Receptor Positive, Her2 Negative Advanced Breast Cancer: Potential Molecular Mechanisms, Clinical Implications and Future Perspectives. Cancers 2021, 13, 332. [Google Scholar] [CrossRef]
- Silvestri, M.; Cristaudo, A.; Morrone, A.; Messina, C.; Bennardo, L.; Nisticò, S.P.; Mariano, M.; Cameli, N. Emerging Skin Toxicities in Patients with Breast Cancer Treated with New Cyclin-Dependent Kinase 4/6 Inhibitors: A Systematic Review. Drug Saf. 2021. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Gao, A.C. Drug Resistance in Castration Resistant Prostate Cancer: Resistance Mechanisms and Emerging Treatment Strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar] [PubMed]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Targeting the Androgen Receptor in Triple-Negative Breast Cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Li, Y.; Song, W.; Xu, Y.; Yang, F.; Zhang, W.; Yin, Y.; Guan, X. Antiproliferative Effect of Androgen Receptor Inhibition in Mesenchymal Stem-Like Triple-Negative Breast Cancer. Cell Physiol. Biochem. 2016, 38, 1003–1014. [Google Scholar] [CrossRef]
- Kong, Y.; Qu, F.; Yuan, X.; Yan, X.; Yu, W. Effect of Bicalutamide on the Proliferation and Invasion of Human Triple Negative Breast Cancer MDA-MB-231 Cells. Medicine 2020, 99, e19822. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.; Arteaga, C.L.; et al. PIK3CA Mutations in Androgen Receptor-Positive Triple Negative Breast Cancer Confer Sensitivity to the Combination of PI3K and Androgen Receptor Inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.R.; Yacoub, R.; Taliaferro-Smith, L.; Osunkoya, A.O.; Odero-Marah, V.A.; Liu, T.; Kimbro, K.S.; Sharma, D.; O’Regan, R.M. Reciprocal Regulation of ZEB1 and AR in Triple Negative Breast Cancer Cells. Breast Cancer Res. Treat. 2010, 123, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490–4494. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lau, K.; Hsu, C.; Chen, J.; Lee, C.; Huang, T.; Chen, Y.; Huang, C.; Lin, P.; Tseng, L. Combination of Palbociclib with Enzalutamide shows in Vitro Activity in RB Proficient and Androgen Receptor Positive Triple Negative Breast Cancer Cells. PLoS ONE 2017, 12, e0189007. [Google Scholar] [CrossRef] [Green Version]
- Christenson, J.L.; O’Neill, K.I.; Williams, M.M.; Spoelstra, N.S.; Jones, K.L.; Trahan, G.D.; Reese, J.; Van Patten, E.T.; Elias, A.; Eisner, J.R.; et al. Activity of Combined Androgen Receptor Antagonism and Cell Cycle Inhibition in Androgen Receptor-Positive Triple-Negative Breast Cancer. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Gordon, M.A.; D’Amato, N.C.; Gu, H.; Babbs, B.; Wulfkuhle, J.; Petricoin, E.F.; Gallagher, I.; Dong, T.; Torkko, K.; Liu, B.; et al. Synergy between Androgen Receptor Antagonism and Inhibition of mTOR and HER2 in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Shanle, E.K.; Zhao, Z.; Hawse, J.; Wisinski, K.; Keles, S.; Yuan, M.; Xu, W. Research Resource: Global Identification of Estrogen Receptor Β Target Genes in Triple Negative Breast Cancer Cells. Mol. Endocrinol. 2013, 27, 1762–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, J.M.; Bruinsma, E.S.; Nelson, A.W.; Chernukhin, I.; Carroll, J.S.; Li, Y.; Subramaniam, M.; Suman, V.J.; Negron, V.; Monroe, D.G.; et al. ERβ-Mediated Induction of Cystatins Results in Suppression of TGFβ Signaling and Inhibition of Triple-Negative Breast Cancer Metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9580–E9589. [Google Scholar] [CrossRef] [Green Version]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.; Thomas, C. ERβ Decreases the Invasiveness of Triple-Negative Breast Cancer Cells by Regulating Mutant p53 Oncogenic Function. Oncotarget 2016, 7, 13599–13611. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; McCollum, C.W.; Bondesson, M.; Quinlan, P.; Thompson, A.; Krishnamurthy, S.; et al. ERbeta1 Represses Basal Breast Cancer Epithelial to Mesenchymal Transition by Destabilizing EGFR. Breast Cancer Res. 2012, 14, R148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Dey, P.; Ziegler, Y.; Jiao, X.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Contrasting Activities of Estrogen Receptor Beta Isoforms in Triple Negative Breast Cancer. Breast Cancer Res. Treat. 2021, 185, 281–292. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.K.; et al. Nuclear and Cytoplasmic Expression of ERbeta1, ERbeta2, and ERbeta5 Identifies Distinct Prognostic Outcome for Breast Cancer Patients. Clin. Cancer Res. 2008, 14, 5228–5235. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, U.K.; Oturkar, C.C.; Adams, C.; Wickramasekera, N.; Bansal, S.; Medisetty, R.; Miller, A.; Swetzig, W.M.; Silwal-Pandit, L.; Børresen-Dale, A.; et al. TP53 Status as a Determinant of Pro- Vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1202–1215. [Google Scholar] [CrossRef]
- Thomas, C.G.; Strom, A.; Lindberg, K.; Gustafsson, J. Estrogen Receptor Beta Decreases Survival of p53-Defective Cancer Cells After DNA Damage by Impairing G₂/M Checkpoint Signaling. Breast Cancer Res. Treat. 2011, 127, 417–427. [Google Scholar] [CrossRef]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and Knockdown of Estrogen Receptor Β Differentially Affect Invasion of Triple-Negative Breast Cancer Cells in Vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef] [Green Version]
- Hinsche, O.; Girgert, R.; Emons, G.; Gründker, C. Estrogen Receptor Β Selective Agonists Reduce Invasiveness of Triple-Negative Breast Cancer Cells. Int. J. Oncol. 2015, 46, 878–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anestis, A.; Sarantis, P.; Theocharis, S.; Zoi, I.; Tryfonopoulos, D.; Korogiannos, A.; Koumarianou, A.; Xingi, E.; Thomaidou, D.; Kontos, M.; et al. Estrogen Receptor Beta Increases Sensitivity to Enzalutamide in Androgen Receptor-Positive Triple-Negative Breast Cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tang, L.; Xu, Y.; Sun, Q.; Yang, F.; Guan, X. ERβ1 Inhibits Metastasis of Androgen Receptor-Positive Triple-Negative Breast Cancer by Suppressing ZEB1. J. Exp. Clin. Cancer Res. 2017, 36, 75. [Google Scholar] [CrossRef] [Green Version]
- McNamara, K.M.; Oguro, S.; Omata, F.; Kikuchi, K.; Guestini, F.; Suzuki, K.; Yang, Y.; Abe, E.; Hirakawa, H.; Brown, K.A.; et al. The Presence and Impact of Estrogen Metabolism on the Biology of Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2017, 161, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O.; Schüler-Toprak, S.; Ortmann, O. Estrogen Actions in Triple-Negative Breast Cancer. Cells 2020, 9, 2358. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, F.; Zhang, W.; Song, W.; Liu, Y.; Guan, X. The Androgen Receptor Promotes Cellular Proliferation by Suppression of G-Protein Coupled Estrogen Receptor Signaling in Triple-Negative Breast Cancer. Cell Physiol. Biochem. 2017, 43, 2047–2061. [Google Scholar] [CrossRef]
- Xu, M.; Yuan, Y.; Yan, P.; Jiang, J.; Ma, P.; Niu, X.; Ma, S.; Cai, H.; Yang, K. Prognostic Significance of Androgen Receptor Expression in Triple Negative Breast Cancer: A Systematic Review and Meta-Analysis. Clin. Breast Cancer 2020, 20, e385–e396. [Google Scholar] [CrossRef] [Green Version]
- Honma, N.; Ogata, H.; Yamada, A.; Matsuda, Y.; Kontani, K.; Miyashita, M.; Arai, T.; Sasaki, E.; Shibuya, K.; Mikami, T.; et al. Clinicopathological Characteristics and Prognostic Marker of Triple-Negative Breast Cancer in Older Women. Hum. Pathol. 2021, 111, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, H.; Han, G.; Pinnaduwage, D.; Murphy, L.C.; Yang, X.R.; Andrulis, I.L.; Sherman, M.; Figueroa, J.; Rimm, D.L. ERβ Splice Variant Expression in Four Large Cohorts of Human Breast Cancer Patient Tumors. Breast Cancer Res. Treat. 2014, 146, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Thike, A.A.; Ong, C.C.H.; Lim, J.X.; Md Nasir, N.D.; Li, H.; Koh, V.C.Y.; Chen, X.; Yeong, J.P.S.; Sasano, H.; et al. Characteristics, Behaviour and Role of Biomarkers in Metastatic Triple-Negative Breast Cancer. J. Clin. Pathol. 2020, 73, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential Response to Neoadjuvant Chemotherapy among 7 Triple-Negative Breast Cancer Molecular Subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Grellety, T.; Callens, C.; Richard, E.; Briaux, A.; Vélasco, V.; Pulido, M.; Gonçalves, A.; Gestraud, P.; MacGrogan, G.; Bonnefoi, H.; et al. Enhancing Abiraterone Acetate Efficacy in Androgen Receptor-Positive Triple-Negative Breast Cancer: Chk1 as a Potential Target. Clin. Cancer Res. 2019, 25, 856–867. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effects | ER-β | GPER |
|---|---|---|
| Agonists | Isoflavone Daidzein Genistein Flavanone Liquiritigenin Prinaberel (ERB-041) Diarylpropionitrile (DPN) WAY200070 | G1 |
| Antagonists | PHTPP | G15 G36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honma, N.; Matsuda, Y.; Mikami, T. Carcinogenesis of Triple-Negative Breast Cancer and Sex Steroid Hormones. Cancers 2021, 13, 2588. https://doi.org/10.3390/cancers13112588
Honma N, Matsuda Y, Mikami T. Carcinogenesis of Triple-Negative Breast Cancer and Sex Steroid Hormones. Cancers. 2021; 13(11):2588. https://doi.org/10.3390/cancers13112588
Chicago/Turabian StyleHonma, Naoko, Yoko Matsuda, and Tetuo Mikami. 2021. "Carcinogenesis of Triple-Negative Breast Cancer and Sex Steroid Hormones" Cancers 13, no. 11: 2588. https://doi.org/10.3390/cancers13112588
APA StyleHonma, N., Matsuda, Y., & Mikami, T. (2021). Carcinogenesis of Triple-Negative Breast Cancer and Sex Steroid Hormones. Cancers, 13(11), 2588. https://doi.org/10.3390/cancers13112588