
Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers

 ,
,  , , and
, , and 
Abstract
:Simple Summary
Abstract
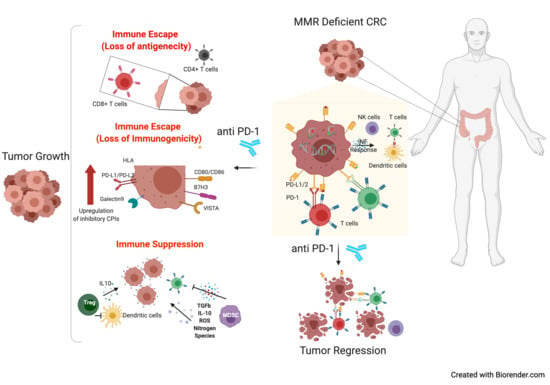
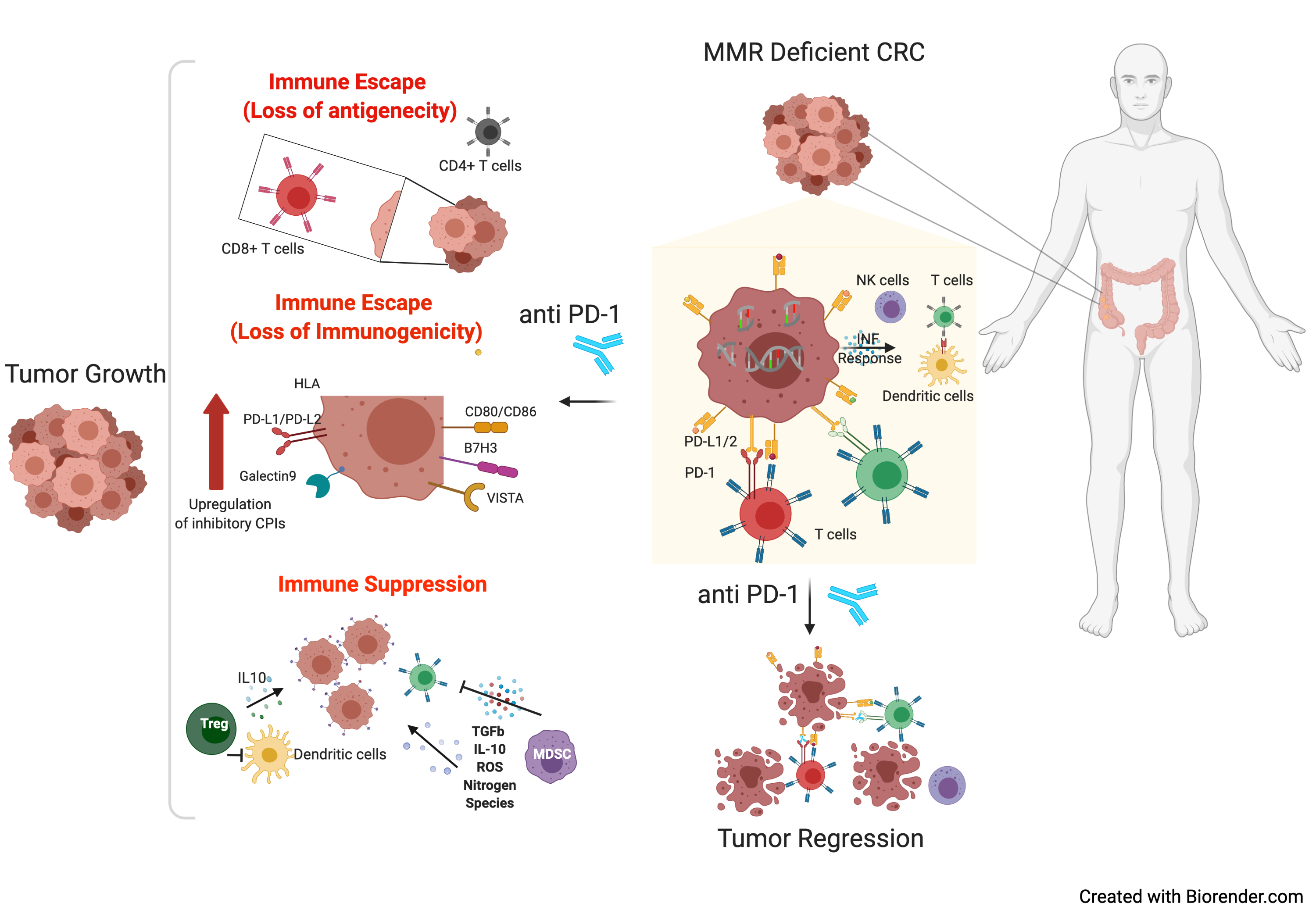
1. Introduction
1.1. The Role of the Gut Microbiota in the Initiation and Progression of MSI CRC
1.2. CRC MMR Deficiency and Immune Surveillance
1.3. Mutational Characteristics of Mismatch-Repair-Deficient Cancer Cells
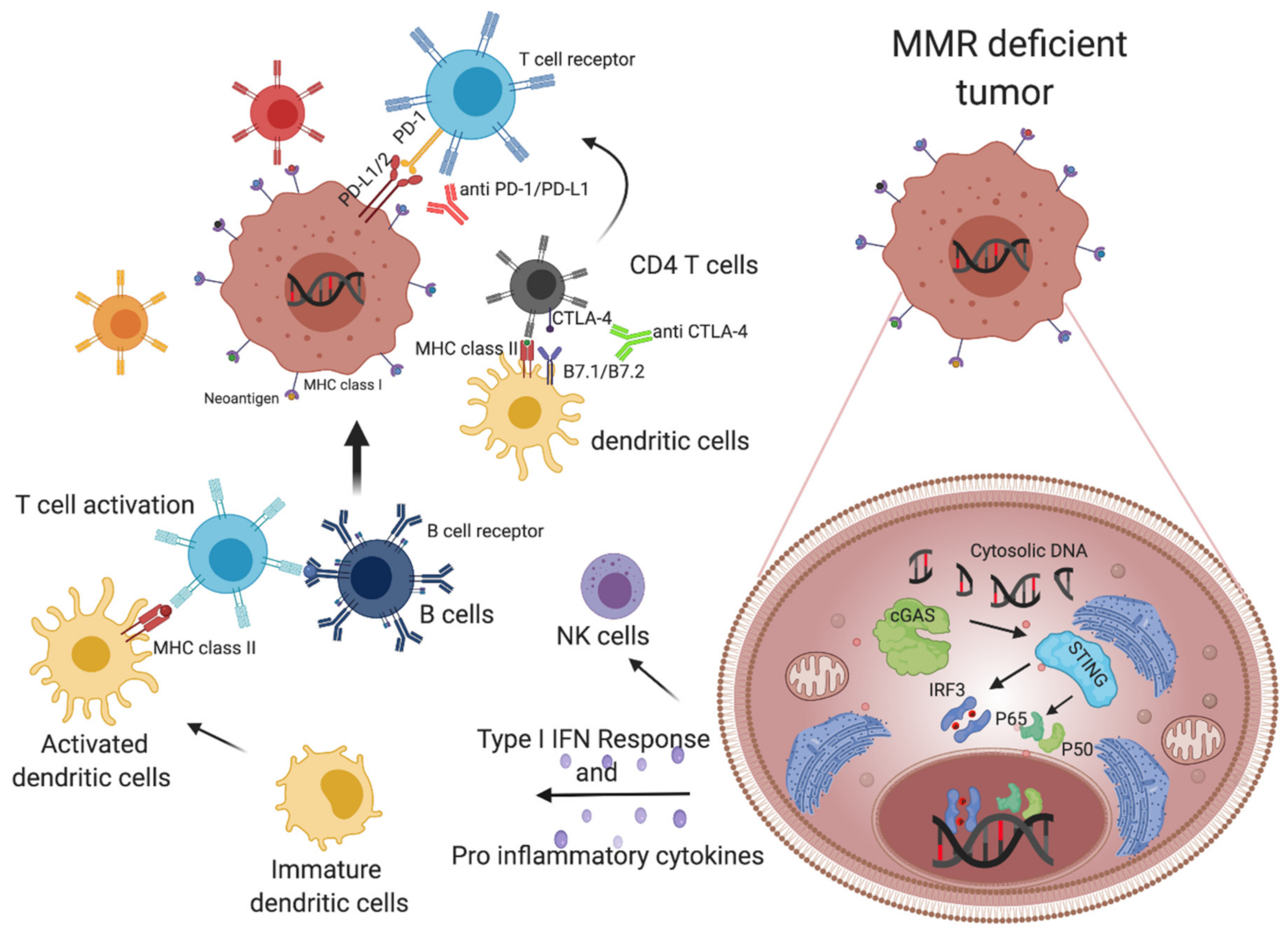
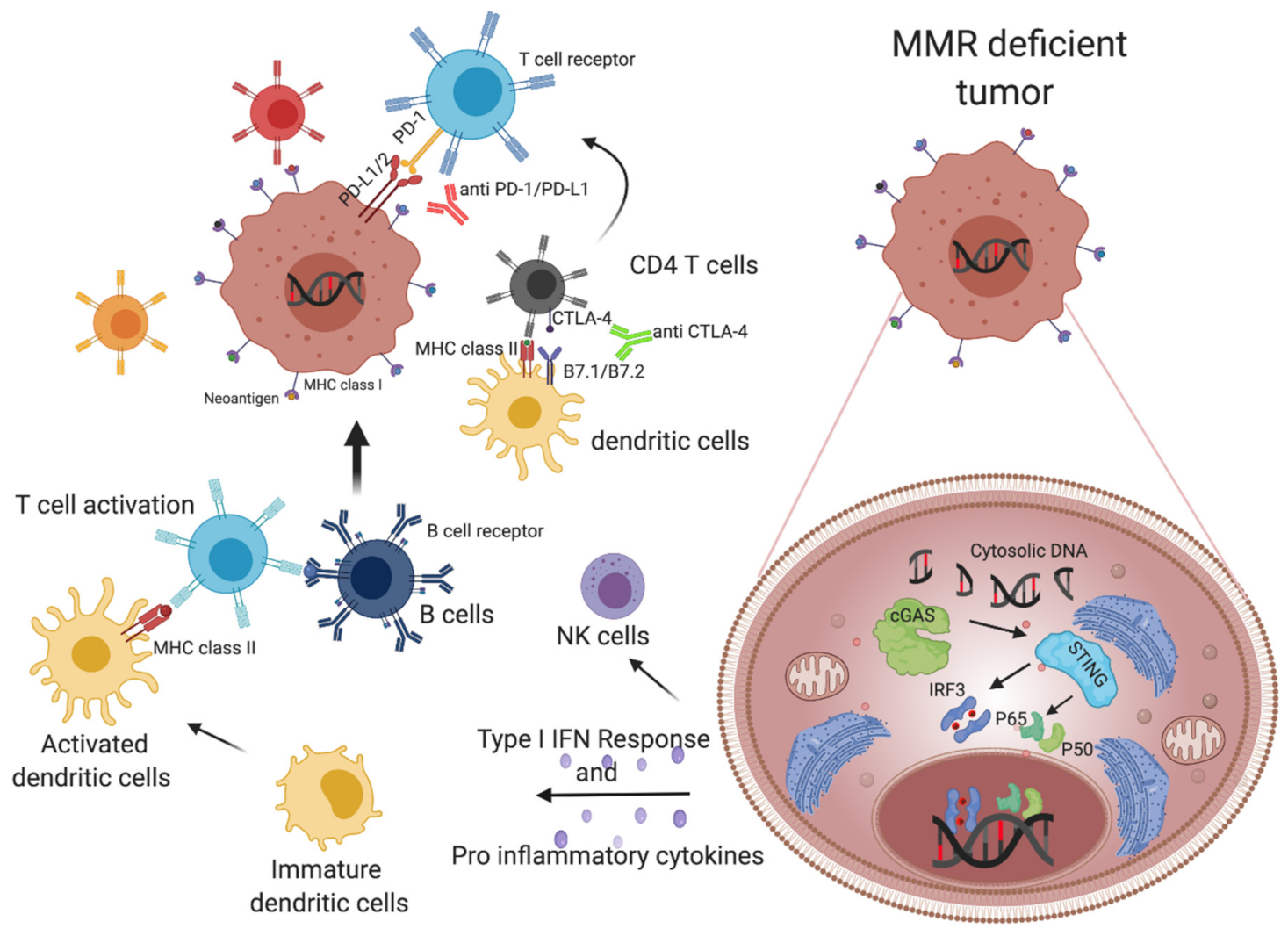
1.4. MMR Deficient Tumors Alert the Immune System, Triggering Cytotoxic T Cells and Inducing an INF-Mediated Immune Response
1.4.1. Neoantigen-Dependent Activation of Immune Surveillance in MMR-Deficient Colorectal Cancers
1.4.2. Cytosolic DNA Release Contributes to the Immunogenic Properties of MMRd Tumors
1.5. The Role of Checkpoint Inhibitor Treatment in MSI mCRC Patients
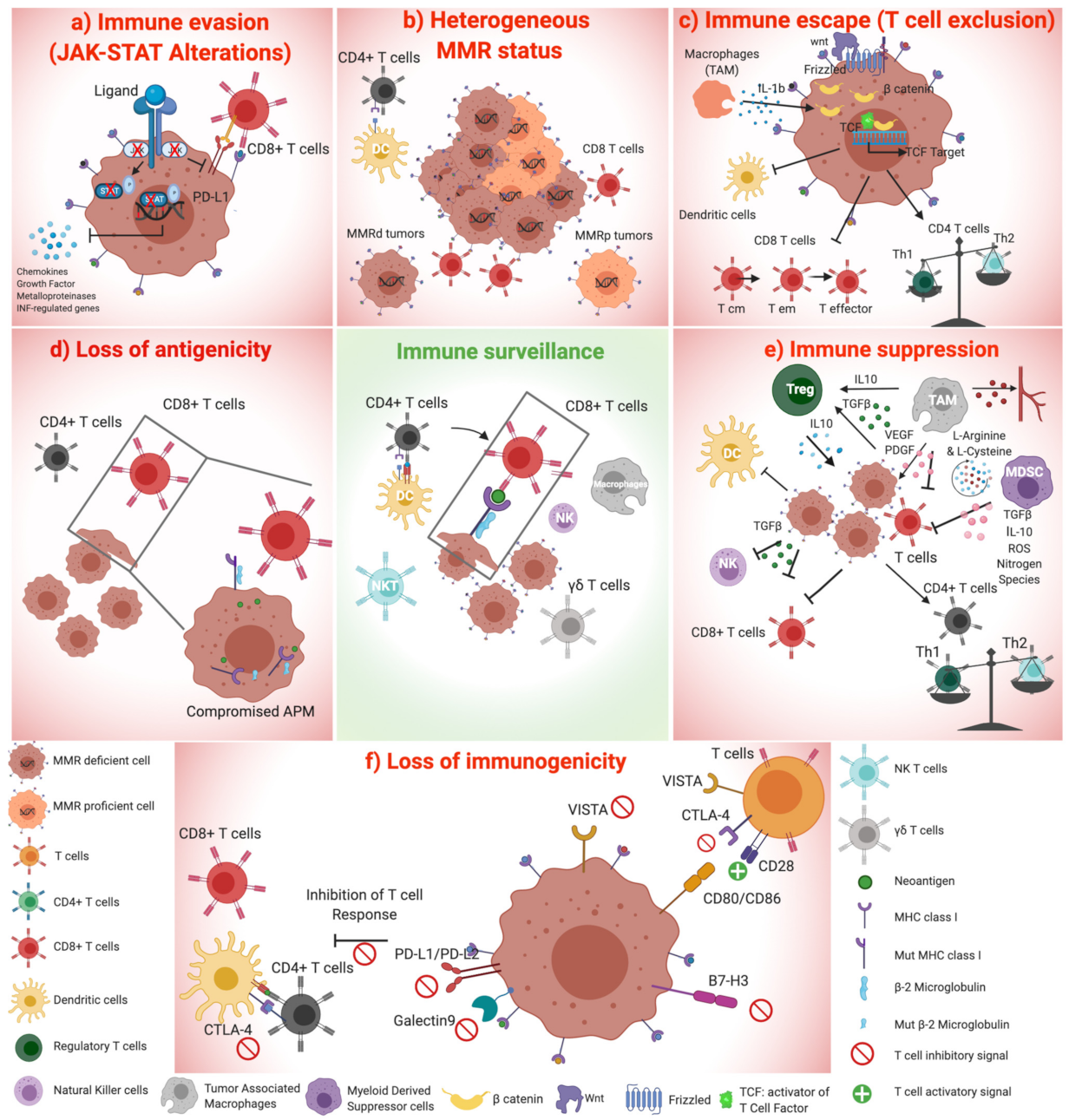
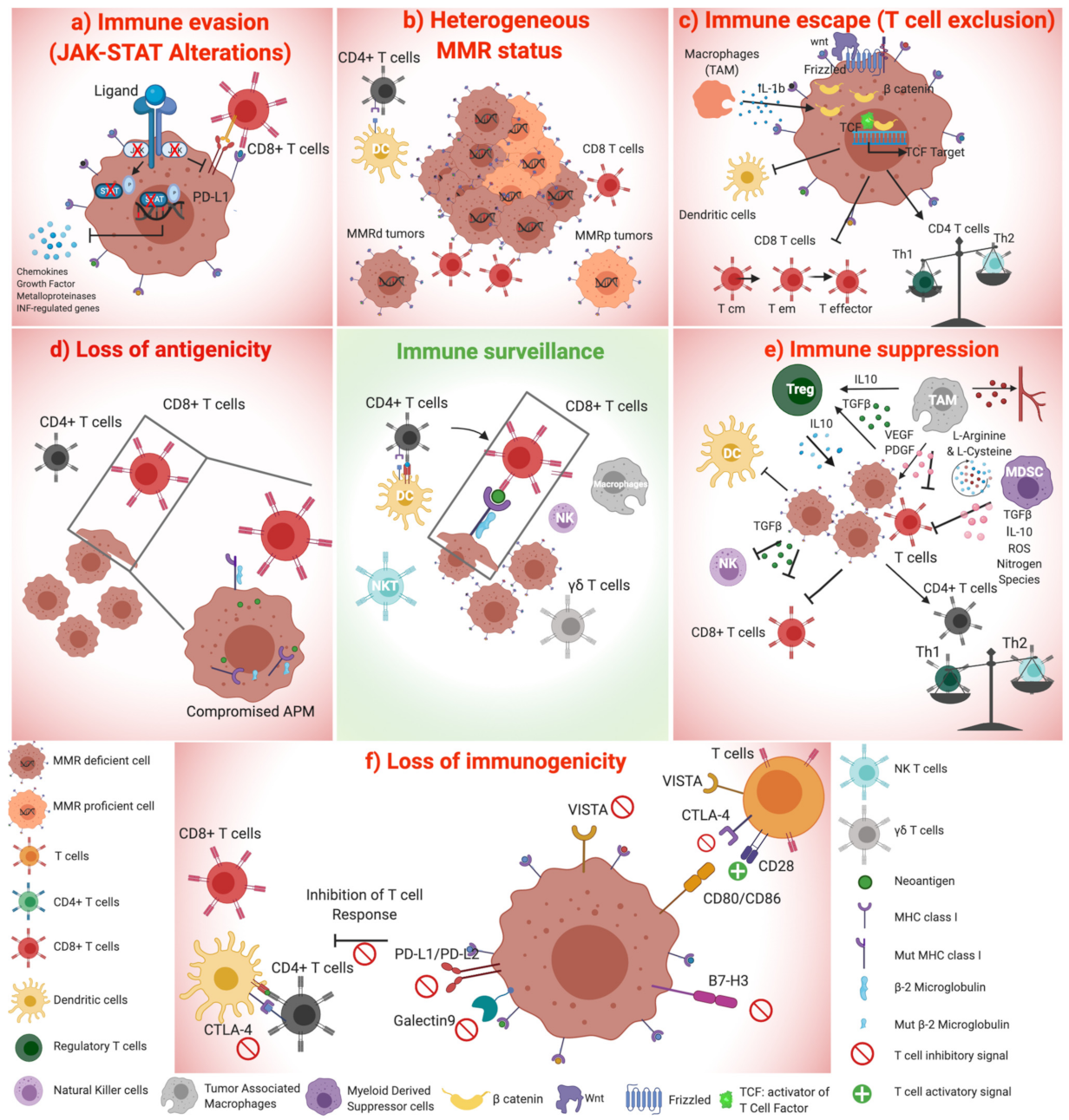
2. Non-Genetic Mechanisms of Immune Evasion in Microsatellite Unstable CRC
2.1. Altered Expression of Inhibitory Immune Checkpoints
2.2. Cytokines, Chemokines, and Factors Orchestrate an Immune Suppressive Microenvironment in MSI Cancer
2.3. cGas-STING Pathway May Alter the Immunogenicity of Cancer Cells and Favor an Immune Suppressive Microenvironment
2.4. Suppressive Immune Cell Compartments in MSI CRC
2.4.1. Regulatory T Cells
2.4.2. The Role of Myeloid-Derived Suppressor Cells
2.4.3. Tumor Associated Macrophages
3. Genetic Mechanisms of Immune Evasion in MSI CRC
3.1. Antigen Presenting Machinery Disruption in MSI Tumors
3.2. Deregulation of WNT Signalling Pathway Alters Tumor Microenvironment, Causing T-Cell Exclusion
3.3. Alterations in JAK-STAT Pathway Orchestrate Cancer Immune Evasion
3.4. Germline Genetic Variants Affect Different Immunomodulatory Pathways
3.5. The Intra-Tumoral Genetic Diversity of MMR Status as a Mechanism of Immune Escape
4. Translational Implications
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Lexical of Abbreviations
| 5-FU | 5-fluorouracil |
| APC | Antigen presenting cell |
| APM | Antigen presenting machinery |
| B2M | β 2 microglobulin |
| BMMR-D | Biallelic mismatch repair deficiency syndrome |
| CCL | Chemokine (cc motif) ligand |
| cGAMP | Cyclic guanosine monophosphate-adenosine monophosphate |
| c-GAS | Cyclic GMP-AMP synthase |
| CIMP | CpG island methylator phenotype |
| CMS | Consensus molecular subtype |
| CN-LOH | Copy-neutral loss of heterozygosity |
| CPIs | Checkpoint inhibitors |
| CRC | Colorectal cancer |
| CSF1-R | Colony stimulating factor 1 receptor |
| CTLA4 | Cytotoxic T-lymphocyte antigen 4 |
| CTNNB1 | β -catenin |
| CXC | Chemokine (cxc motif) |
| CXCL | Chemokine (cxc motif) ligand |
| DBS | Double base substitution |
| DC | Dendritic cell |
| DCR | Disease control rate |
| DFCI | Dana Farber Cancer Institute |
| DOR | Duration of response |
| ENPP1 | Ectonucleotide pyrophosphatase/phosphodiesterase 1 |
| ERP57 | Endoplasmic reticulum protein 57 |
| EXO 1 | Exonuclease 1 |
| FOXP 3 | Forkhead box 3 |
| FS | Frameshift |
| GATA3 | GATA binding protein 3 |
| GROα | Growth regulated protein α |
| GSK3B | Glycogen synthase kinase 3 beta |
| HLA | Human leukocyte antigen |
| HNPCC | Hereditary non-polyposis colorectal cancer |
| HPV | Human Papilloma Virus |
| ICOS | Inducible T-cell costimulator |
| IDO | Indoleamine 2,3-dioxygenase |
| IL- | Interleukin- |
| IL1RA | IL-1 receptor agonist |
| IN/DEL | Insertion/deletion |
| INF | Interferon |
| IRF3 | Interferon regulatory factor 3 |
| JAK | Janus kinases |
| LAG3 | Lymphocyte activation gene 3 |
| M1 | M1-like macrophages |
| M2 | M2-like macrophages |
| mCRC | Metastatic CRC |
| MDSC | Myeloid derived suppressor cells |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MHC | Major histocompatibility complex |
| MLH1 | MutL homolog 1 |
| MMR | Mismatch repair |
| MMRd | Mismatch repair deficient |
| MMRp | Mismatch repair proficient |
| mPFS | Median progression free survival |
| MSH2 | MutS homolog 2 |
| MSH6 | MutS homolog 6 |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NFKB | Nuclear factor Kappa-ligand-chain-enhancer of activated B cells |
| NGS | Next generation sequencing |
| NK | Natural killer |
| NKG2D | Natural killer group 2 member D |
| NLRC5 | NLR family CARD domain containing 5 |
| NSCLC | Non-small cell lung cancer |
| ORR | Objective response rate |
| OS | Overall survival |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death protein ligand 1 |
| PFS | Progression free survival |
| PMS2 | PMS1 homolog 2 |
| POLD1 | Polymerase delta 1 |
| POLE | Polymerase epsilon |
| RAE1 | Retinoic acid early transcript 1 ligand |
| RCC | Renal cell carcinoma |
| RFX5 | Regulatory factor X5 |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| SBS | Single base substitution |
| SNVs | Single nucleotide variants |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon gene |
| TAM | Tumor associated macrophages |
| TAP | Transporter associated with antigen processing |
| TCF4 | T cell factor 4 |
| TCGA | The Cancer Genome Atlas |
| TCR | T cell receptor |
| Tfh | T follicular helper |
| TGFBR2 | TGF-β receptor 2 |
| TGF-β | Transforming growth factor β |
| Th1 | T-helper type 1 |
| TIM3 | T-cell immunoglobulin and mucin domain-3 |
| TMB | Tumor mutational burden |
| Treg | Regulatory T cells |
| VEGF | Vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T cell activation |
References
- Michielin, O.; Van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metge, J.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- van Dijk, N.; Gil-Jimenez, A.; Silina, K.; Hendricksen, K.; Smit, L.A.; de Feijter, J.M.; van Montfoort, M.L.; van Rooijen, C.; Peters, D.; Broeks, A.; et al. Preoperative ipilimumab plus nivolumab in locoregionally advanced urothelial cancer: The NABUCCO trial. Nat. Med. 2020, 26, 1839–1844. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.-J.; Oaknin, A.; Kerger, J.; López-Picazo, J.M.; Machiels, J.-P.; Delord, J.-P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Seock-Ah, I.; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA A Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef]
- Adam, R.; Kitano, Y. Multidisciplinary approach of liver metastases from colorectal cancer. Ann. Gastroenterol. Surg. 2019, 3, 50–56. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; Van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Sargent, D.J. Molecular Pathways: Microsatellite Instability in Colorectal Cancer: Prognostic, Predictive, and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.-P.; Markowitz, S.; Willson, J.K.V.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Venderbosch, S.; Nagtegaal, I.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [Green Version]
- Benatti, P.; Gafà, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite Instability and Colorectal Cancer Prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [Green Version]
- Tougeron, D.; Sueur, B.; Zaanan, A.; De La Fouchardiére, C.; Sefrioui, D.; LeComte, T.; Aparicio, T.; Guetz, G.D.; Artru, P.; Hautefeuille, V.; et al. Prognosis and chemosensitivity of deficient MMR phenotype in patients with metastatic colorectal cancer: An AGEO retrospective multicenter study. Int. J. Cancer 2020, 147, 285–296. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, J.L.A. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Hendler, R.; Zhang, Y. Probiotics in the Treatment of Colorectal Cancer. Medicines 2018, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.B. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ 2017, 356, j831. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 1–7. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Honda, K. Intestinal Commensal Microbes as Immune Modulators. Cell Host Microbe 2012, 12, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host–microbiota maladaptation in colorectal cancer. Nat. Cell Biol. 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Weisburger, J.H.; Reddy, B.S.; Narisawa, T.; Wynder, E.L. Germ-Free Status and Colon Tumor Induction by N-Methyl-N’-Nitro-N-Nitrosoguanidine. Exp. Biol. Med. 1975, 148, 1119–1121. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [Green Version]
- Hofseth, L.J.; Hebert, J.R.; Chanda, A.; Chen, H.; Love, B.L.; Pena, M.M.; Murphy, E.A.; Sajish, M.; Sheth, A.; Buckhaults, P.J.; et al. Publisher Correction: Early-onset colorectal cancer: Initial clues and current views. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 517. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2011, 22, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Temraz, S.; Nassar, F.; Nasr, R.; Charafeddine, M.; Mukherji, D.; Shamseddine, A. Gut Microbiome: A Promising Biomarker for Immunotherapy in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4155. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Zhang, X.; Mima, K.; Bullman, S.; Sukawa, Y.; Nowak, J.A.; Kosumi, K.; Masugi, Y.; Twombly, T.S.; Cao, Y.; et al. Fusobacterium nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability Status. Cancer Immunol. Res. 2018, 6, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Welch, J.L.M.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2019, 30, 2012. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, J.-F.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti–PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef]
- Miller, P.L.; Carson, T.L. Mechanisms and microbial influences on CTLA-4 and PD-1-based immunotherapy in the treatment of cancer: A narrative review. Gut Pathog. 2020, 12, 1–10. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Derosa, L.; Hellmann, M.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.; Arbour, K.; Chaft, J.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- Yoshino, T.; Arnold, D.; Taniguchi, H.; Pentheroudakis, G.; Yamazaki, K.; Xu, R.-H.; Kim, T.; Ismail, F.; Tan, I.; Yeh, K.-H.; et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO–ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann. Oncol. 2018, 29, 44–70. [Google Scholar] [CrossRef]
- Becht, E.; De Reyniès, A.; Giraldo, N.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar] [PubMed]
- Guidoboni, M.; Gafà, R.; Viel, A.; Doglioni, C.; Russo, A.; Santini, A.; Del Tin, L.; Macrì, E.; Lanza, G.; Boiocchi, M.; et al. Microsatellite Instability and High Content of Activated Cytotoxic Lymphocytes Identify Colon Cancer Patients with a Favorable Prognosis. Am. J. Pathol. 2001, 159, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Huyghe, N.; Baldin, P.; van den Eynde, M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: What is the future beyond deficient mismatch-repair tumours? Gastroenterol. Rep. 2020, 8, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Al-Shibli, K.I.; Donnem, T.; Al-Saad, S.; Persson, M.; Bremnes, R.M.; Busund, L.-T. Prognostic Effect of Epithelial and Stromal Lymphocyte Infiltration in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, K.; Miyamoto, M.; Cho, Y.; Suzuoki, M.; Oshikiri, T.; Nakakubo, Y.; Itoh, T.; Ohbuchi, T.; Kondo, S.; Katoh, H. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br. J. Cancer 2006, 94, 275–280. [Google Scholar] [CrossRef]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Zhang, J.; Luo, P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front. Immunol. 2020, 11, 2039. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Gulubova, M.V.; Ananiev, J.R.; Vlaykova, T.I.; Yovchev, Y.; Tsoneva, V.; Manolova, I.M. Role of dendritic cells in progression and clinical outcome of colon cancer. Int. J. Color. Dis. 2011, 27, 159–169. [Google Scholar] [CrossRef]
- Dadabayev, A.R.; Sandel, M.H.; Menon, A.G.; Morreau, H.; Melief, C.J.M.; Offringa, R.; Van Der Burg, S.H.; Rhijn, C.J.-V.; Ensink, N.G.; Tollenaar, R.A.E.M.; et al. Dendritic cells in colorectal cancer correlate with other tumor-infiltrating immune cells. Cancer Immunol. Immunother. 2004, 53, 978–986. [Google Scholar] [CrossRef]
- Bauer, K.; Michel, S.; Reuschenbach, M.; Nelius, N.; Doeberitz, M.V.K.; Kloor, M. Dendritic cell and macrophage infiltration in microsatellite-unstable and microsatellite-stable colorectal cancer. Fam. Cancer 2011, 10, 557–565. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durno, C.A.; Sherman, P.M.; Aronson, M.; Malkin, D.; Hawkins, C.; Bakry, D.; Bouffet, E.; Gallinger, S.; Pollett, A.; Campbell, B.; et al. Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur. J. Cancer 2015, 51, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. 2021, 11, 1176–1191. [Google Scholar]
- Germano, G.; Amirouchene-Angelozzi, N.; Rospo, G.; Bardelli, A. The Clinical Impact of the Genomic Landscape of Mismatch Repair–Deficient Cancers. Cancer Discov. 2018, 8, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021, 39, 96–108.e6. [Google Scholar] [CrossRef]
- Rospo, G.; Lorenzato, A.; Amirouchene-Angelozzi, N.; Magrì, A.; Cancelliere, C.; Corti, G.; Negrino, C.; Amodio, V.; Montone, M.; Bartolini, A.; et al. Evolving neoantigen profiles in colorectal cancers with DNA repair defects. Genome Med. 2019, 11, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nat. Cell Biol. 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Nakayama, M. Antigen Presentation by MHC-Dressed Cells. Front. Immunol. 2014, 5, 672. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- Bjerregaard, A.-M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017, 66, 1123–1130. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 1–21. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope Landscape in Breast and Colorectal Cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Schreiber, R.D. The odds of immunotherapy success. Science 2015, 350, 158–159. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res. 2020, 80, 3972–3982. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’Donnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.-L.; Blank, S.V.; et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell 2020, 183, 1634–1649.e17. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Sanchez, A.H.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Laumont, C.M.; Daouda, T.; Laverdure, J.-P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.-P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef]
- He, L.; Chen, Y.; Wu, Y.; Xu, Y.; Zhang, Z.; Liu, Z. Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell. Mol. Life Sci. 2017, 74, 2395–2411. [Google Scholar] [CrossRef]
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, H.D.P.; Mohamed, E.; Huang, L.; Ou, R.; Pacholczyk, G.; Arbab, A.S.; Munn, D.; Mellor, A.L. STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res. 2016, 76, 2076–2081. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nat. Cell Biol. 2016, 533, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Xu, T.; Cui, M. cGAS-STING signaling in cancer immunity and immunotherapy. Biomed. Pharmacother. 2021, 133, 110972. [Google Scholar] [CrossRef]
- Gerlinger, M. Immunotherapy Sensitivity of Mismatch Repair-Deficient Cancer: Mutation Load Is Not Enough. Cancer Cell 2021, 39, 16–18. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Hu, J.; Zhang, H.; Xu, F.; He, W.; Wang, X.; Li, M.; Lu, W.; Zeng, G.; et al. cGAS/STING axis mediates a topoisomerase II inhibitor–induced tumor immunogenicity. J. Clin. Investig. 2019, 129, 4850–4862. [Google Scholar] [CrossRef]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763.e4. [Google Scholar] [CrossRef] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.R.; Le Bert, N.; Ho, S.S.; Shen, Y.J.; Tang, M.L.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 Ligands for the NKG2D Receptor Are Regulated by STING-Dependent DNA Sensor Pathways in Lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef] [Green Version]
- Takashima, K.; Takeda, Y.; Oshiumi, H.; Shime, H.; Okabe, M.; Ikawa, M.; Matsumoto, M.; Seya, T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 2016, 478, 1764–1771. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e5. [Google Scholar] [CrossRef]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]
- Lenz, H.-J. Nivolumab (NIVO) + low-dose ipilimumab (IPI) as first-line (1L) therapy in microsatellite instability-high/DNA mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): Clinical update. J. Clin. Oncol. 2019, 38. [Google Scholar] [CrossRef]
- Lenz, H.-J. Subgroup analyses of patients (pts) with microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC) treated with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line (1L) therapy:Two-year clinical update. J. Clin. Oncol. 2021, 39 (Suppl. 3), 58. [Google Scholar]
- Cohen, R.; Colle, R.; Pudlarz, T.; Heran, M.; Duval, A.; Svrcek, M.; André, T. Immune Checkpoint Inhibition in Metastatic Colorectal Cancer Harboring Microsatellite Instability or Mismatch Repair Deficiency. Cancers 2021, 13, 1149. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Berg, J.G.V.D.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Hain, E.; Buhard, O.; Guilloux, A.; Bardier, A.; Kaci, R.; Bertheau, P.; Renaud, F.; Bibeau, F.; Fléjou, J.-F.; et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer With Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncol. 2019, 5, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Depetris, I.; Biason, P.; Intini, R.; Prete, A.A.; Leone, F.; Lombardi, P.; Filippi, R.; Spallanzani, A.; Cascinu, S.; et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes. Oncologist 2020, 25, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.; Guan, R.; Yang, H.; Zhou, Y.; Hong, W.; Ma, L.; Zhao, G.; Yu, M. Assessment of the expression of the immune checkpoint molecules PD-1, CTLA4, TIM-3 and LAG-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int. J. Cancer 2020, 147, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune Checkpoint Modulation in Colorectal Cancer: What’s New and What to Expect. J. Immunol. Res. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Saito, H.; Ikeguchi, M. Upregulation of TIM-3 and PD-1 on CD4+ and CD8+ T Cells Associated with Dysfunction of Cell-Mediated Immunity after Colorectal Cancer Operation. Yonago Acta Medica 2012, 55, 1–9. [Google Scholar]
- Fabrizio, D.A.; Jr, T.J.G.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Kloor, M.; Doeberitz, M.V.K. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [Green Version]
- VanderWalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Metzger, T.C.; Long, H.; Potluri, S.; Pertel, T.; Bailey-Bucktrout, S.L.; Lin, J.C.; Fu, T.; Sharma, P.; Allison, J.P.; Feldman, R.M. ICOS Promotes the Function of CD4+ Effector T Cells during Anti-OX40–Mediated Tumor Rejection. Cancer Res. 2016, 76, 3684–3689. [Google Scholar] [CrossRef] [Green Version]
- Boissière-Michot, F.; Lazennec, G.; Frugier, H.; Jarlier, M.; Roca, L.; Duffour, J.; Du Paty, E.; Laune, D.; Blanchard, F.; Le Pessot, F.; et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. OncoImmunology 2014, 3, e29256. [Google Scholar] [CrossRef] [Green Version]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer 2004, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Rubie, C.; Frick, V.O.; Wagner, M.; Schuld, J.; Gräber, S.; Brittner, B.; Bohle, R.M.; Schilling, M.K. ELR+ CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer 2008, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Yuan, A.; Goll, R.; Vonen, B.; Florholmen, J. Dynamic changes of interleukin-8 network along the colorectal adenoma–carcinoma sequence. Cancer Immunol. Immunother. 2009, 58, 1897–1905. [Google Scholar] [CrossRef]
- Jiang, Z.; Xu, Y.; Cai, S. CXCL10 expression and prognostic significance in stage II and III colorectal cancer. Mol. Biol. Rep. 2009, 37, 3029–3036. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Alevizopoulos, A.; Mermod, N. Transforming growth factor-beta: The breaking open of a black box. Bioessays 1997, 19, 581–591. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.T.; Gorham, J.D. TGF-beta 1 regulates antigen-specific CD4+ T cell responses in the periphery. J. Immunol. 2007, 179, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Bevan, M.J. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012, 13, 667–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sad, S.; Mosmann, T.R. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J. Immunol. 1994, 153, 3514–3522. [Google Scholar] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [Green Version]
- Nandan, D.; Reiner, N.E. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J. Immunol. 1997, 158, 1095–1101. [Google Scholar]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.-J.; Park, S.H. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun. 2011, 2, 460. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lim, S.; Li, A.G.; Lee, C.; Lee, Y.S.; Lee, E.-K.; Park, S.H.; Wang, X.-J.; Kim, S.-J. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat. Immunol. 2007, 8, 504–513. [Google Scholar] [CrossRef]
- de Miranda, N.F. Transforming Growth Factor β Signaling in Colorectal Cancer Cells With Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology 2015, 148, 1427–1437e.8. [Google Scholar] [CrossRef] [Green Version]
- Warusavitarne, J. Restoring TGFbeta function in microsatellite unstable (MSI-H) colorectal cancer reduces tumourigenicity but increases metastasis formation. Int. J. Colorectal. Dis. 2009, 24, 139–144. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Liang, D.; Xiao-Feng, H.; Guan-Jun, D.; Er-Ling, H.; Sheng, C.; Ting-Ting, W.; Qin-Gang, H.; Yan-Hong, N.; Ya-Yi, H. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2494–2503. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Konno, H.; Barber, G.N. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene 2015, 34, 5302–5308. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Vareki, S.M. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Truini, A.; Germano, G.; Bardelli, A. Inactivation of DNA repair—prospects for boosting cancer immune surveillance. Genome Med. 2018, 10, 91. [Google Scholar] [CrossRef]
- Puccini, A.; Battaglin, F.; Iaia, M.L.; Lenz, H.-J.; Salem, M.E. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J. Immunother. Cancer 2019, 8, e000404. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Waniczek, D.; Lorenc, Z.; Śnietura, M.; Wesecki, M.; Kopec, A.; Muc-Wierzgoń, M. Tumor-Associated Macrophages and Regulatory T Cells Infiltration and the Clinical Outcome in Colorectal Cancer. Arch. Immunol. Ther. Exp. 2017, 65, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Kuwahara, T.; Hazama, S.; Suzuki, N.; Yoshida, S.; Tomochika, S.; Nakagami, Y.; Matsui, H.; Shindo, Y.; Kanekiyo, S.; Tokumitsu, Y.; et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br. J. Cancer 2019, 121, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Cavalleri, T.; Bianchi, P.; Basso, G.; Celesti, G.; Grizzi, F.; Bossi, P.; Greco, L.; Pitrone, C.; Valtorta, E.; Mauri, G.; et al. Combined Low Densities of FoxP3+ and CD3+ Tumor-Infiltrating Lymphocytes Identify Stage II Colorectal Cancer at High Risk of Progression. Cancer Immunol. Res. 2019, 7, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Le Gouvello, S.; Bastuji-Garin, S.; Aloulou, N.; Mansour, H.; Chaumette, M.-T.; Berrehar, F.; Seikour, A.; Charachon, A.; Karoui, M.; Leroy, K.; et al. High prevalence of Foxp3 and IL17 in MMR-proficient colorectal carcinomas. Gut 2008, 57, 772–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, S.; Benner, A.; Tariverdian, M.; Wentzensen, N.; Hoefler, P.; Pommerencke, T.; Grabe, N.; Doeberitz, M.V.K.; Kloor, M. High density of FOXP3-positive T cells infiltrating colorectal cancers with microsatellite instability. Br. J. Cancer 2008, 99, 1867–1873. [Google Scholar] [CrossRef] [Green Version]
- Byrne, W.L.; Mills, K.; Lederer, J.A.; O’Sullivan, G.C. Targeting Regulatory T Cells in Cancer: Figure 1. Cancer Res. 2011, 71, 6915–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, T.-H.; Yang, F.-F.; Zhu, Y.-X.; Li, Y.-L.; Lei, Q.; Song, X.-J.; Xia, Y.; Xiong, Y.; Zhang, L.-D.; Wang, N.-Y.; et al. Inhibition of Stat3 signaling pathway by nifuroxazide improves antitumor immunity and impairs colorectal carcinoma metastasis. Cell Death Dis. 2018, 8, e2534. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572e.7. [Google Scholar] [CrossRef] [Green Version]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef]
- Mantovani, A.; Locati, M. Tumor-Associated Macrophages as a Paradigm of Macrophage Plasticity, Diversity, and Polarization. Arter. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Forssell, J.; Öberg, Å.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High Macrophage Infiltration along the Tumor Front Correlates with Improved Survival in Colon Cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.; Kawaguchi, T.; Peng, X.; Qi, Q.; Liu, S.; Yan, L.; Takabe, K. Tumor Infiltrating Lymphocytes and Macrophages Improve Survival in Microsatellite Unstable Colorectal Cancer. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2010, 4, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Yang, Y.; Qi, L.; Chen, J.; Ge, W.; Zheng, S. Subtyping of microsatellite instability-high colorectal cancer. Cell Commun. Signal. 2019, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Hajjami, H.M.-E.; et al. T cell–induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B. CSF1R. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nat. Cell Biol. 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Mantovani, A.; Allavena, P. Targeting of the innate immunity/inflammation as complementary anti-tumor therapies. Ann. Med. 2011, 43, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Sveen, A.; Johannessen, B.; Tengs, T.; Danielsen, S.A.; Eilertsen, I.A.; Lind, G.E.; Berg, K.C.G.; Leithe, E.; Meza-Zepeda, L.A.; Domingo, E.; et al. Multilevel genomics of colorectal cancers with microsatellite instability—clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Kim, T.-M.; Laird, P.W.; Park, P.J. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Sayaman, R.W.; Saad, M.; Thorsson, V.; Hu, D.; Hendrickx, W.; Roelands, J.; Porta-Pardo, E.; Mokrab, Y.; Farshidfar, F.; Kirchhoff, T.; et al. Germline genetic contribution to the immune landscape of cancer. Immunity 2021, 54, 367–386.e8. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Samstein, R.M.; Lee, K.-W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.-H.; Liu, Y.-J.; Bayir, E.; Iliopoulos, D.; Elsen, P.V.D.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [Green Version]
- Meissner, T.B.; Liu, Y.-J.; Lee, K.-H.; Li, A.; Biswas, A.; Van Eggermond, M.C.J.A.; Elsen, P.V.D.; Kobayashi, K.S. NLRC5 Cooperates with the RFX Transcription Factor Complex To Induce MHC Class I Gene Expression. J. Immunol. 2012, 188, 4951–4958. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, M.; Janikovits, J.; Doeberitz, M.V.K.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. OncoImmunology 2018, 7, e1445453. [Google Scholar] [CrossRef] [Green Version]
- Kloor, M.; Michel, S.; Doeberitz, M.V.K. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef]
- Van Kaer, L. Major histocompatibility complex classI-restricted antigen processing and presentation. Tissue Antigens 2002, 60, 1–9. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of Functional Beta2-Microglobulin in Metastatic Melanomas From Five Patients Receiving Immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Middha, S.; Yaeger, R.; Shia, J.; Stadler, Z.K.; King, S.; Guercio, S.; Paroder, V.; Bates, D.D.; Rana, S.; Diaz, L.A.; et al. Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and Are Microsatellite Instability-High. JCO Precis. Oncol. 2019, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lu, S.; Rospo, G.; Lamba, S.; Rousseau, B.; Fanelli, S.; Stenech, D.; Le, D.T.; Hays, J.; Totaro, M.G.; et al. CD4 T cell dependent rejection of beta 2 microglobulin null mismatch repair deficient tumors. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nat. Cell Biol. 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Sharma, A.; Oh, S.Y.; Moon, H.; Hossain, M.Z.; Salay, T.M.; Leeds, K.E.; Du, H.; Wu, B.; Waterman, M.L.; et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat. Immunol. 2009, 10, 992–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsberry, W.N.; Meza-Perez, S.; Londoño, A.I.; Katre, A.A.; Mott, B.T.; Roane, B.M.; Goel, N.; Wall, J.A.; Cooper, S.J.; Norian, L.A.; et al. Inhibiting WNT Ligand Production for Improved Immune Recognition in the Ovarian Tumor Microenvironment. Cancers 2020, 12, 766. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Tinsley, H.N.; Keetona, A.; Qua, Z.; Piazza, G.A.; Lia, Y. Suppression of Wnt/beta-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2009, 602, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Gowda, P. Mutant Isocitrate Dehydrogenase 1 Disrupts PKM2-β-Catenin-BRG1 Transcriptional Network-Driven CD47 Expression. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Schürch, C.M.; Forster, S.; Brühl, F.; Yang, S.H.; Felley-Bosco, E.; Hewer, E. The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. OncoImmunology 2018, 7, e1373235. [Google Scholar] [CrossRef]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [Green Version]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [Green Version]
- Di Piazza, M.; Nowell, C.S.; Koch, U.; Durham, A.-D.; Radtke, F. Loss of Cutaneous TSLP-Dependent Immune Responses Skews the Balance of Inflammation from Tumor Protective to Tumor Promoting. Cancer Cell 2012, 22, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Gheidari, F.; Bakhshandeh, B.; Teimoori-Toolabi, L.; Mehrtash, A.; Ghadir, M.; Zeinali, S. TCF4 silencing sensitizes the colon cancer cell line to oxaliplatin as a common chemotherapeutic drug. Anti-Cancer Drugs 2014, 25, 908–916. [Google Scholar] [CrossRef]
- Kukcinaviciute, E.; Jonusiene, V.; Sasnauskiene, A.; Dabkeviciene, D.; Eidenaite, E.; Laurinavicius, A. Significance of Notch and Wnt signaling for chemoresistance of colorectal cancer cells HCT116. J. Cell. Biochem. 2018, 119, 5913–5920. [Google Scholar] [CrossRef]
- Kim, W.K.; Byun, W.S.; Chung, H.-J.; Oh, J.; Park, H.J.; Choi, J.S.; Lee, S.K. Esculetin suppresses tumor growth and metastasis by targeting Axin2/E-cadherin axis in colorectal cancer. Biochem. Pharmacol. 2018, 152, 71–83. [Google Scholar] [CrossRef]
- Heim, M.; Kerr, I.; Stark, G.; Darnell, J. Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science 1995, 267, 1347–1349. [Google Scholar] [CrossRef]
- Albacker, L.A.; Wu, J.; Smith, P.; Warmuth, M.; Stephens, P.J.; Zhu, P.; Yu, L.; Chmielecki, J. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS ONE 2017, 12, e0176181. [Google Scholar] [CrossRef]
- Dawson, N.A.; Zibelman, M.; Lindsay, T.; Feldman, R.A.; Saul, M.; Gatalica, Z.; Korn, W.M.; Heath, E.I. An Emerging Landscape for Canonical and Actionable Molecular Alterations in Primary and Metastatic Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Robert, D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nat. Cell Biol. 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immun. 2020, 52, 55–81. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Capo-Chichi, J.; Spence, T.; Grenier, S.; Stockley, T.; Kamel-Reid, S.; Serra, S.; Sabatini, P.; Chetty, R. Heterogenous loss of mismatch repair (MMR) protein expression: A challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J. Pathol. Clin. Res. 2019, 5, 115–129. [Google Scholar] [CrossRef]
- Fusco, N.; Lopez, G.; Corti, C.; Pesenti, C.; Colapietro, P.; Ercoli, G.; Gaudioso, G.; Faversani, A.; Gambini, D.; Michelotti, A.; et al. Mismatch Repair Protein Loss as a Prognostic and Predictive Biomarker in Breast Cancers Regardless of Microsatellite Instability. JNCI Cancer Spectr. 2018, 2, pky056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapusot, C.; Martin, L.; Bouvier, A.M.; Bonithon-Kopp, C.; Ecarnot-Laubriet, A.; Rageot, D.; Ponnelle, T.; Laurent-Puig, P.; Faivre, J.; Piard, F. Microsatellite instability and intratumoural heterogeneity in 100 right-sided sporadic colon carcinomas. Br. J. Cancer 2002, 87, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, A.; Kariv, R.; Solar, I.; Hershkovitz, D. Geographic Heterogeneity for Mismatch Repair Proteins Is Associated with Defects in DNA Repair. Isr. Med Assoc. J. 2020, 22, 32–36. [Google Scholar] [PubMed]
- Loupakis, F.; Maddalena, G.; Depetris, I.; Murgioni, S.; Bergamo, F.; Tos, A.P.D.; Rugge, M.; Munari, G.; Nguyen, A.; Szeto, C.; et al. Treatment with checkpoint inhibitors in a metastatic colorectal cancer patient with molecular and immunohistochemical heterogeneity in MSI/dMMR status. J. Immunother. Cancer 2019, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- He, W.-Z.; Hu, W.-M.; Wang, F.; Rong, Y.-M.; Yang, L.; Xie, Q.-K.; Yang, Y.-Z.; Jiang, C.; Qiu, H.-J.; Lu, J.-B.; et al. Comparison of Mismatch Repair Status Between Primary and Matched Metastatic Sites in Patients With Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 1174–1183. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hong, D.S. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Mauri, G.; Bonazzina, E.; Amatu, A.; Tosi, F.; Bencardino, K.; Gori, V.; Massihnia, D.; Cipani, T.; Spina, F.; Ghezzi, S.; et al. The Evolutionary Landscape of Treatment for BRAFV600E Mutant Metastatic Colorectal Cancer. Cancers 2021, 13, 137. [Google Scholar] [CrossRef]
- Cocoran, R.; Giannakis, M.; Allen, J.; Chen, J.; Pelka, K.; Chao, S.; Meyerhardt, J.; Enzinger, A.; Enzinger, P.; McCleary, N.; et al. Yugelun M Clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. Ann. Oncol. 2020, 31. [Google Scholar] [CrossRef]
- Turgeon, G.; Weickhardt, A.; Azad, A.A.; Solomon, B.; Siva, S. Radiotherapy and immunotherapy: A synergistic effect in cancer care. Med. J. Aust. 2018, 210, 47–53. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Clinical Strategy | Trial Identifier | Phase | Regimen |
|---|---|---|---|
| CPIs combinations | NCT04008030 (CheckMate 8HW) | III | Nivolumab and ipilimumab vs. standard cytotoxic regimens |
| CPIs plus targeted agents - RAS inhibitors - BRAF inhibitors | NCT03785249 (Krystal 1) | I/II | MRTX849 and pembrolizumab |
| NCT04185883 (Codebreak) | Ib/II | Sotorasib + PD1i | |
| NCT03668431 | II | Dabrafenib + trametinib + spartalizumab | |
| NCT04294160 | Ib | Dabrafenib + LTT462 (ERKi) + Spartalizumab (PDR001) | |
| CPIs plus cytotoxic and/or anti-VEGF agents | NCT02997228 (COMMIT) | III | FOLFOX + bevacizumab + atezolizumab vs. atezolizumab monotherapy vs. FOLFOX + bevacizumab |
| CPIs plus radiotherapy | NCT04001101 | II | Pembrolizumab ± RT |
| NCT03104439 | II | Nivolumab + ipilimumab + RT | |
| Converting strategy | NCT03519412 (ARETHUSA) | II | Temozolomide followed by pembrolizumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amodio, V.; Mauri, G.; Reilly, N.M.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A.; Germano, G. Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers 2021, 13, 2638. https://doi.org/10.3390/cancers13112638
Amodio V, Mauri G, Reilly NM, Sartore-Bianchi A, Siena S, Bardelli A, Germano G. Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers. 2021; 13(11):2638. https://doi.org/10.3390/cancers13112638
Chicago/Turabian StyleAmodio, Vito, Gianluca Mauri, Nicole M. Reilly, Andrea Sartore-Bianchi, Salvatore Siena, Alberto Bardelli, and Giovanni Germano. 2021. "Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers" Cancers 13, no. 11: 2638. https://doi.org/10.3390/cancers13112638
APA StyleAmodio, V., Mauri, G., Reilly, N. M., Sartore-Bianchi, A., Siena, S., Bardelli, A., & Germano, G. (2021). Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers, 13(11), 2638. https://doi.org/10.3390/cancers13112638