
Chromosomal Instability in Acute Myeloid Leukemia

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
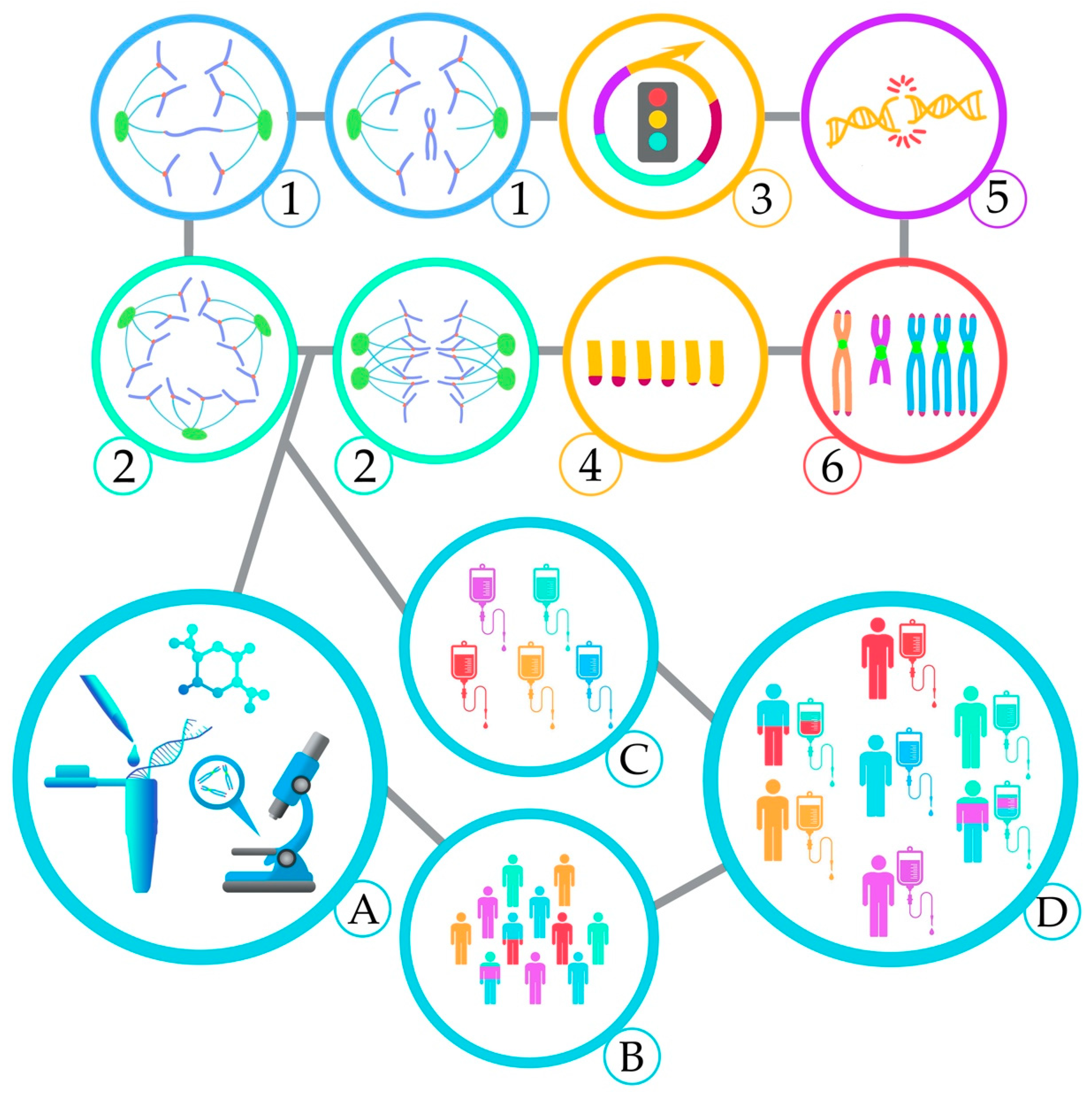
2. Mechanisms and Consequences of CIN
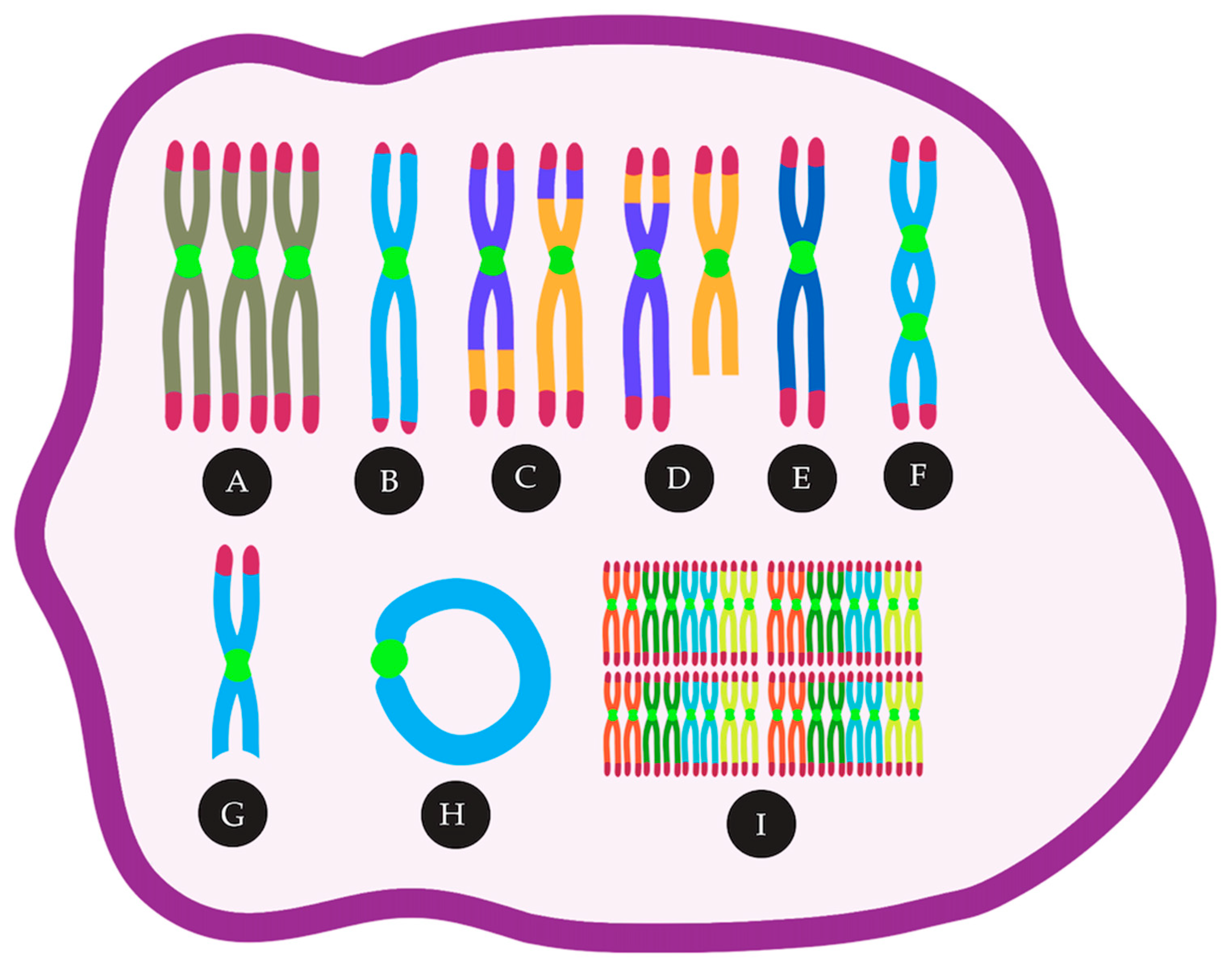
2.1. Aneuploidy and CIN in AML, Mechanism and Consequence — An Inter-Relationship
2.2. Chromosome Segregation Errors
2.2.1. Defects in the Spindle Assembly Checkpoint (SAC)
2.2.2. Cohesion Defects
2.2.3. Centrosome Dysfunction and Assembly of Multipolar Mitotic Spindles
2.3. DNA Double-strand Breaks
2.4. Telomere Dysfunction
2.5. Complex Chromosomal Rearrangements
2.6. Epigenetic Regulation
3. Clinical Considerations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-Damage Response during Mitosis Induces Whole-Chromosome Missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef]
- Targa, A.; Rancati, G. Cancer: A CINful evolution. Curr. Opin. Cell Biol. 2018, 52, 136–144. [Google Scholar] [CrossRef]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Genet. 1999, 15, M57–M60. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal Instability Confers Intrinsic Multidrug Resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Zhang, W.; Sood, A.K. Chromosomal Instability in Tumor Initiation and Development. Cancer Res. 2019, 79, 3995–4002. [Google Scholar] [CrossRef]
- Salgueiro, L.; Buccitelli, C.; Rowald, K.; Somogyi, K.; Kandala, S.; Korbel, J.O.; Sotillo, R. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef]
- Hoevenaar, W.H.M.; Janssen, A.; Quirindongo, A.I.; Ma, H.; Klaasen, S.J.; Teixeira, A.; van Gerwen, B.; Lansu, N.; Morsink, F.H.M.; Offerhaus, G.J.A.; et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 2020, 11, 1501. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Fam, H.K.; Wang, Y.K.; Styles, E.B.; Kim, J.-H.; Ang, J.S.; Singh, T.; Larionov, V.; Shah, S.P.; Andrews, B.; et al. Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 9967–9976. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Villegas, V.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Hulegårdh, E.; Nilsson, C.; Lazarevic, V.; Garelius, H.; Antunovic, P.; Rangert Derolf, Å.; Möllgård, L.; Uggla, B.; Wennström, L.; Wahlin, A.; et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: A report from the Swedish Acute Leukemia Registry. Am. J. Hematol. 2015, 90, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Sill, H.; Olipitz, W.; Zebisch, A.; Schulz, E.; Wölfler, A. Therapy-related myeloid neoplasms: Pathobiology and clinical characteristics. Br. J. Pharmacol. 2011, 162, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in acute leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Grimwade, D.; Mrózek, K. Diagnostic and Prognostic Value of Cytogenetics in Acute Myeloid Leukemia. Hematol. Oncol. Clin. 2011, 25, 1135–1161. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Jin, N.; Lera, R.F.; Yan, R.E.; Guo, F.; Oxendine, K.; Horner, V.L.; Hu, Y.; Wan, J.; Mattison, R.J.; Weaver, B.A.; et al. Chromosomal instability upregulates interferon in acute myeloid leukemia. Genes Chromosom. Cancer 2020, gcc.22880. [Google Scholar] [CrossRef]
- Mrózek, K. Cytogenetic, Molecular Genetic, and Clinical Characteristics of Acute Myeloid Leukemia with a Complex Karyotype. Semin. Oncol. 2008, 35, 365–377. [Google Scholar] [CrossRef]
- Fröhling, S.; Schlenk, R.F.; Kayser, S.; Morhardt, M.; Benner, A.; Döhner, K.; Döhner, H. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: Results from AMLSG trial AML HD98-B. Blood 2006, 108, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Archer, K.J.; Mrózek, K.; Ruppert, A.S.; Carroll, A.J.; Vardiman, J.W.; Pettenati, M.J.; Baer, M.R.; Qumsiyeh, M.B.; Koduru, P.R.; et al. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: Results from Cancer and Leukemia Group B 8461. Blood 2006, 108, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Van Der Holt, B.; Breems, D.A.; Berna Beverloo, H.; Van Den Berg, E.; Burnett, A.K.; Sonneveld, P.; Löwenberg, B. Various distinctive cytogenetic abnormalities in patients with acute myeloid leukaemia aged 60 years and older express adverse prognostic value: Results from a prospective clinical trial. Br. J. Haematol. 2007, 136, 96–105. [Google Scholar] [CrossRef]
- Schoch, C.; Haferlach, T.; Haase, D.; Fonatsch, C.; Loffler, H.; Schlegelberger, B.; Staib, P.; Sauerland, M.C.; Heinecke, A.; Buchner, T.; et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: A study of 90 patients. Br. J. Haematol. 2001, 112, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): Analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001, 98, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cheng, Y.; Wang, B. Cancer and Genomic Instability. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 463–486. [Google Scholar]
- Compton, D.A. Mechanisms of aneuploidy. Curr. Opin. Cell Biol. 2011, 23, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Schukken, K.M.; Foijer, F. CIN and Aneuploidy: Different Concepts, Different Consequences. BioEssays 2018, 40, 1700147. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Hitzler, J.K.; Zipursky, A. Origins of leukaemia in children with Down syndrome. Nat. Rev. Cancer 2005, 5, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ganmore, I.; Smooha, G.; Izraeli, S. Constitutional aneuploidy and cancer predisposition. Hum. Mol. Genet. 2009, 18, R84–R93. [Google Scholar] [CrossRef]
- Breems, D.A.; Van Putten, W.L.J.; De Greef, G.E.; Van Zelderen-Bhola, S.L.; Gerssen-Schoorl, K.B.J.; Mellink, C.H.M.; Nieuwint, A.; Jotterand, M.; Hagemeijer, A.; Beverloo, H.B.; et al. Monosomal Karyotype in Acute Myeloid Leukemia: A Better Indicator of Poor Prognosis Than a Complex Karyotype. J. Clin. Oncol. 2008, 26, 4791–4797. [Google Scholar] [CrossRef]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.; Archer, K.; Mrozek, K.; Vardiman, J.; Carroll, A.; Pettenati, M.; Moore, J.; Kolitz, J.; Mayer, R.; Stone, R.; et al. Isolated trisomy of chromosomes 8, 11, 13 and 21 is an adverse prognostic factor in adults with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B 8461. Int. J. Oncol. 2002, 21, 1041–1051. [Google Scholar] [CrossRef]
- Herold, T.; Metzeler, K.H.; Vosberg, S.; Hartmann, L.; Röllig, C.; Stölzel, F.; Schneider, S.; Hubmann, M.; Zellmeier, E.; Ksienzyk, B.; et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood 2014, 124, 1304–1311. [Google Scholar] [CrossRef]
- Perrot, A.; Luquet, I.; Pigneux, A.; Mugneret, F.; Delaunay, J.; Harousseau, J.-L.; Barin, C.; Cahn, J.-Y.; Guardiola, P.; Himberlin, C.; et al. Dismal prognostic value of monosomal karyotype in elderly patients with acute myeloid leukemia: A GOELAMS study of 186 patients with unfavorable cytogenetic abnormalities. Blood 2011, 118, 679–685. [Google Scholar] [CrossRef]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Linke, S.P.; Paulson, T.G.; Huang, L.C. Maintaining genetic stability through TP53 mediated checkpoint control. Cancer Surv. 1997, 29, 183–219. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Blount, P.L.; Galipeau, P.C.; Sanchez, C.A.; Neshat, K.; Levine, D.S.; Yin, J.; Suzuki, H.; Abraham, J.M.; Meltzer, S.J.; Reid, B.J. 17p allelic losses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Res. 1994, 54, 2292–2295. [Google Scholar]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef]
- Reinisch, A.; Chan, S.M.; Thomas, D.; Majeti, R. Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin. Hematol. 2015, 52, 150–164. [Google Scholar] [CrossRef]
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood 2017, 129, 2587–2591. [Google Scholar] [CrossRef]
- Fukasawa, K.; Wiener, F.; Woude, G.F.V.; Mai, S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene 1997, 15, 1295–1302. [Google Scholar] [CrossRef]
- Dalton, W.B.; Yu, B.; Yang, V.W. p53 suppresses structural chromosome instability after mitotic arrest in human cells. Oncogene 2010, 29, 1929–1940. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef]
- Tan, B.X.; Khoo, K.H.; Lim, T.M.; Lane, D.P. High Mdm4 levels suppress p53 activity and enhance its half-life in acute myeloid leukaemia. Oncotarget 2014, 5, 933–943. [Google Scholar] [CrossRef]
- Li, L.; Tan, Y.; Chen, X.; Xu, Z.; Yang, S.; Ren, F.; Guo, H.; Wang, X.; Chen, Y.; Li, G.; et al. MDM4 Overexpressed in Acute Myeloid Leukemia Patients with Complex Karyotype and Wild-Type TP53. PLoS ONE 2014, 9, e113088. [Google Scholar] [CrossRef]
- Bueso-Ramos, C.; Yang, Y.; DeLeon, E.; McCown, P.; Stass, S.; Albitar, M. The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.-Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16 INK4a /p14 ARF locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, A.; Schlegel, C.; Jansen, I.; Bochtler, T.; Jauch, A.; Krämer, A. TP53 deficiency permits chromosome abnormalities and karyotype heterogeneity in acute myeloid leukemia. Leukemia 2019, 33, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Spielberger, R.T.; Thangavelu, M.; Zhao, N.; Davis, E.M.; Iannantuoni, K.; Larson, R.A.; Le Beau, M.M. dic(5;17): A recurring abnormality in malignant myeloid disorders associated with mutations ofTP53. Genes Chromosom. Cancer 1997, 20, 282–291. [Google Scholar] [CrossRef]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations with Loss of Heterozygosity of p53 Are Common in Therapy-Related Myelodysplasia and Acute Myeloid Leukemia After Exposure to Alkylating Agents and Significantly Associated With Deletion or Loss of 5q, a Complex Karyotype, and a Poor Prognosis. J. Clin. Oncol. 2001, 19, 1405–1413. [Google Scholar] [CrossRef]
- Reilly, A.; Busch, S.; Abkowitz, J.L.; Becker, P.S.; Doulatov, S. Deletions of 5q Promote Genome Instability in TP53-Mutant MDS/AML. Blood 2019, 134, 1244. [Google Scholar] [CrossRef]
- Schoch, C.; Haferlach, T.; Bursch, S.; Gerstner, D.; Schnittger, S.; Dugas, M.; Kern, W.; Löffler, H.; Hiddemann, W. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: A detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Genes Chromosom. Cancer 2002, 35, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.N.; Johnson, P.R.E.; Moorman, A.V.; Roman, E.; Willett, E.; Taylor, P.R.; Proctor, S.J.; Bown, N.; Ogston, S.; Bowen, D.T. Population-based demographic study of karyotypes in 1709 patients with adult Acute Myeloid Leukemia. Leukemia 2006, 20, 444–450. [Google Scholar] [CrossRef]
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosom. Cancer 2014, 53, 402–410. [Google Scholar] [CrossRef]
- Jin, N.; Burkard, M. Abstract 3797: Chromosome instability as a prognostic factor an immunotherapeutic target in acute myeloid leukemia. In Proceedings of the Immunology, American Association for Cancer Research. Chicago, IL, USA, 14–18 April 2018; p. 3797. [Google Scholar]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.V.S.; Benezra, R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; Foltz, D.R.; Cleveland, D.W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 8699–8704. [Google Scholar] [CrossRef]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.V.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Ghatak, D.; Das, P.; Roychoudhury, S. Transcriptional Control of Mitosis: Deregulation and Cancer. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, X. Correlation of defective mitotic checkpoint with aberrantly reduced expression of MAD2 protein in nasopharyngeal carcinoma cells. Carcinogenesis 2000, 21, 2293–2297. [Google Scholar] [CrossRef]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; FitzPatrick, D.; Kidd, A.; Méhes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M.; Blundell, T.L. BUB1 and BUBR1: Multifaceted kinases of the cell cycle. Trends Biochem. Sci. 2011, 36, 141–150. [Google Scholar] [CrossRef]
- Jacquemont, S.; Bocéno, M.; Rival, J.M.; Méchinaud, F.; David, A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. 2002, 109, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Schnerch, D.; Yalcintepe, J.; Schmidts, A.; Becker, H.; Follo, M.; Engelhardt, M.; Wäsch, R. Cell cycle control in acute myeloid leukemia. Am. J. Cancer Res. 2012, 2, 508–528. [Google Scholar]
- Ghelli Luserna di Rorà, A.; Martinelli, G.; Simonetti, G. The balance between mitotic death and mitotic slippage in acute leukemia: A new therapeutic window? J. Hematol. Oncol. 2019, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Nishii, K.; Usui, E.; Katayama, N.; Lorenzo V, F.; Nakase, K.; Kobayashi, T.; Miwa, H.; Mizutani, M.; Tanaka, I.; Nasu, K.; et al. Characteristics of t(8;21) acute myeloid leukemia (AML) with additional chromosomal abnormality: Concomitant trisomy 4 may constitute a distinctive subtype of t(8;21) AML. Leukemia 2003, 17, 731–737. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boyapati, A.; Yan, M.; Peterson, L.F.; Biggs, J.R.; Le Beau, M.M.; Zhang, D.-E. A leukemia fusion protein attenuates the spindle checkpoint and promotes aneuploidy. Blood 2007, 109, 3963–3971. [Google Scholar] [CrossRef]
- Jeganathan, K.B.; Malureanu, L.; van Deursen, J.M. The Rae1–Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature 2005, 438, 1036–1039. [Google Scholar] [CrossRef]
- Salsi, V.; Ferrari, S.; Gorello, P.; Fantini, S.; Chiavolelli, F.; Mecucci, C.; Zappavigna, V. NUP98 Fusion Oncoproteins Promote Aneuploidy by Attenuating the Mitotic Spindle Checkpoint. Cancer Res. 2014, 74, 1079–1090. [Google Scholar] [CrossRef]
- Tanaka, K. Establishment and maintenance of sister chromatid cohesion in fission yeast by a unique mechanism. EMBO J. 2001, 20, 5779–5790. [Google Scholar] [CrossRef]
- Cucco, F.; Musio, A. Genome stability: What we have learned from cohesinopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2016, 172, 171–178. [Google Scholar] [CrossRef]
- Barber, T.D.; McManus, K.; Yuen, K.W.Y.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef]
- Sajesh, B.V.; Lichtensztejn, Z.; McManus, K.J. Sister chromatid cohesion defects are associated with chromosome instability in Hodgkin lymphoma cells. BMC Cancer 2013, 13, 391. [Google Scholar] [CrossRef]
- De Koninck, M.; Losada, A. Cohesin Mutations in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026476. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Bollin, R.; Gehlhaar, M.; Walter, C.; Dugas, M.; Suchanek, K.J.; Kirchner, A.; Huang, L.; Chaturvedi, A.; Wichmann, M.; et al. Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 2014, 123, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Ostronoff, F.; Othus, M.; Lazenby, M.; Estey, E.; Appelbaum, F.R.; Evans, A.; Godwin, J.; Gilkes, A.; Kopecky, K.J.; Burnett, A.; et al. Prognostic Significance of NPM1 Mutations in the Absence of FLT3–Internal Tandem Duplication in Older Patients with Acute Myeloid Leukemia: A SWOG and UK National Cancer Research Institute/Medical Research Council Report. J. Clin. Oncol. 2015, 33, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J. Mutations in AML with a normal karyotype: NPM1 and FLT3-ITD, ready to use as a key prognosticator? Korean J. Hematol. 2010, 45, 79. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Messa, F.; Rosso, V.; Arruga, F.; Defilippi, I.; Carturan, S.; Catalano, R.; Pautasso, M.; Panuzzo, C.; Nicoli, P.; et al. Increase sensitivity to chemotherapeutical agents and cytoplasmatic interaction between NPM leukemic mutant and NF-κB in AML carrying NPM1 mutations. Leukemia 2008, 22, 1234–1240. [Google Scholar] [CrossRef][Green Version]
- Brinkley, B. Managing the centrosome numbers game: From chaos to stability in cancer cell division. Trends Cell Biol. 2001, 11, 18–21. [Google Scholar] [CrossRef]
- Nigg, E.A. Origins and consequences of centrosome aberrations in human cancers. Int. J. Cancer 2006, 119, 2717–2723. [Google Scholar] [CrossRef]
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome Abnormalities and Chromosome Instability Occur Together in Pre-invasive Carcinomas. Cancer Res. 2003, 63, 1398–1404. [Google Scholar]
- Hsu, L.-C.; Kapali, M.; DeLoia, J.A.; Gallion, H.H. Centrosome abnormalities in ovarian cancer. Int. J. Cancer 2005, 113, 746–751. [Google Scholar] [CrossRef]
- Krämer, A.; Neben, K.; Ho, A.D. Centrosome aberrations in hematological malignancies. Cell Biol. Int. 2005, 29, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Pihan, G.A. Centrosome Dysfunction Contributes to Chromosome Instability, Chromoanagenesis, and Genome Reprograming in Cancer. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Neben, K.; Giesecke, C.; Schweizer, S.; Ho, A.D.; Krämer, A. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood 2003, 101, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora A Protein Kinase: To the Centrosome and Beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef]
- Carmena, M.; Ruchaud, S.; Earnshaw, W.C. Making the Auroras glow: Regulation of Aurora A and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 2009, 21, 796–805. [Google Scholar] [CrossRef]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Nobumoto, A.; Udaka, K.; Yokoyama, A. CD34+/CD38− acute myelogenous leukemia cells aberrantly express Aurora kinase A. Int. J. Cancer 2013, 133, 3706–3719. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Garcia-Manero, G.; Kantarjian, H.M.; Xiao, L.; Vadhan-Raj, S.; Fernandez, M.H.; Nguyen, M.H.; Medeiros, L.J.; Bueso-Ramos, C.E. Analysis of Aurora kinase A expression in CD34+ blast cells isolated from patients with myelodysplastic syndromes and acute myeloid leukemia. J. Hematop. 2009, 2, 2–8. [Google Scholar] [CrossRef][Green Version]
- Huang, X.-F.; Luo, S.-K.; Xu, J.; Li, J.; Xu, D.-R.; Wang, L.-H.; Yan, M.; Wang, X.-R.; Wan, X.-B.; Zheng, F.-M.; et al. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood 2008, 111, 2854–2865. [Google Scholar] [CrossRef]
- Lucena-Araujo, A.R.; de Oliveira, F.M.; Leite-Cueva, S.D.; dos Santos, G.A.; Falcao, R.P.; Rego, E.M. High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leuk. Res. 2011, 35, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Aplan, P.D. Chromosomal aberrations induced by double strand DNA breaks. DNA Repair 2005, 4, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhang, Z.; Roschke, A.; Varga, T.; Aplan, P.D. Generation of Gross Chromosomal Rearrangements by a Single Engineered DNA Double Strand Break. Sci. Rep. 2017, 7, 43156. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.O.; Alt, F.W. DNA double strand break repair and chromosomal translocation: Lessons from animal models. Oncogene 2001, 20, 5572–5579. [Google Scholar] [CrossRef]
- Damelin, M.; Bestor, T.H. The decatenation checkpoint. Br. J. Cancer 2007, 96, 201–205. [Google Scholar] [CrossRef]
- Khan, F.A.; Ali, S.O. Physiological Roles of DNA Double-Strand Breaks. J. Nucleic Acids 2017, 2017, 1–20. [Google Scholar] [CrossRef]
- Bower, J.J.; Karaca, G.F.; Zhou, Y.; Simpson, D.A.; Cordeiro-Stone, M.; Kaufmann, W.K. Topoisomerase IIα maintains genomic stability through decatenation G2 checkpoint signaling. Oncogene 2010, 29, 4787–4799. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.S.; Clarke, D.J.; Mullinger, A.M.; Giménez-Abián, J.F.; Creighton, A.M.; Johnson, R.T. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature 1994, 372, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.; Williamson, E.A.; Sheema, S.; Lee, S.-H.; Libby, E.; Willman, C.L.; Nickoloff, J.A.; Hromas, R. Metnase mediates chromosome decatenation in acute leukemia cells. Blood 2009, 114, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, M.A.; De Jesus Pizarro, R.E.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014, 28, 1242–1251. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Kuzyk, A.; Mai, S. c-MYC-Induced Genomic Instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.S.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. MYC Can Induce DNA Breaks In vivo and In vitro Independent of Reactive Oxygen Species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef]
- Li, Z.; Owonikoko, T.K.; Sun, S.-Y.; Ramalingam, S.S.; Doetsch, P.W.; Xiao, Z.-Q.; Khuri, F.R.; Curran, W.J.; Deng, X. c-Myc Suppression of DNA Double-strand Break Repair. Neoplasia 2012, 14, 1190–IN35. [Google Scholar] [CrossRef]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.-J.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [CrossRef]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Hemsing, A.L.; Hovland, R.; Tsykunova, G.; Reikvam, H. Trisomy 8 in acute myeloid leukemia. Expert Rev. Hematol. 2019, 12, 947–958. [Google Scholar] [CrossRef]
- Jaff, N.; Chelghoum, Y.; Elhamri, M.; Tigaud, I.; Michallet, M.; Thomas, X. Trisomy 8 as sole anomaly or with other clonal aberrations in acute myeloid leukemia: Impact on clinical presentation and outcome. Leuk. Res. 2007, 31, 67–73. [Google Scholar] [CrossRef]
- Vaniawala, S.N.; Patel, M.V.; Chavda, P.D.; Zaveri, S.H.; Gadhia, P.K. The possible significance of trisomy 8 in acute myeloid leukemia. Int. J. Res. Med. Sci. 2017, 5, 2652. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef]
- De Lange, T. How Telomeres Solve the End-Protection Problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.L.; Hirose, Y.; Langmore, J.P. Long G Tails at Both Ends of Human Chromosomes Suggest a C Strand Degradation Mechanism for Telomere Shortening. Cell 1997, 88, 657–666. [Google Scholar] [CrossRef]
- Mattarocci, S.; D’Ambrosio, E.; Tafaro, L.; Somma, V.; Zannino, G.; Marigliano, V.; Ascenzioni, F.; Cimino-Reale, G. Erosion of telomeric 3′-overhangs in white blood cells of aged subjects with high frequency of very short telomeres. Mech. Ageing Dev. 2011, 132, 27–32. [Google Scholar] [CrossRef]
- Calado, R.T.; Regal, J.A.; Kajigaya, S.; Young, N.S. Erosion of telomeric single-stranded overhang in patients with aplastic anaemia carrying telomerase complex mutations. Eur. J. Clin. Investig. 2009, 39, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Han, B.; Wu, Y.; Zhou, D.; Zhao, Y. Telomerase gene mutation screening and telomere overhang detection in Chinese patients with acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Schaefer, K.-L.; Jauch, A.; Keller, M.; Wai, D.; Brinkschmidt, C.; van Valen, F.; Boecker, W.; Dockhorn-Dworniczak, B.; Poremba, C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001, 20, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Aalbers, A.M.; Calado, R.T.; Young, N.S.; Zwaan, C.M.; Wu, C.; Kajigaya, S.; Coenen, E.A.; Baruchel, A.; Geleijns, K.; de Haas, V.; et al. Telomere length and telomerase complex mutations in pediatric acute myeloid leukemia. Leukemia 2013, 27, 1786–1789. [Google Scholar] [CrossRef]
- Mosrati, M.A.; Willander, K.; Falk, I.J.; Hermanson, M.; Höglund, M.; Stockelberg, D.; Wei, Y.; Lotfi, K.; Söderkvist, P. Association between TERT promoter polymorphisms and acute myeloid leukemia risk and prognosis. Oncotarget 2015, 6, 25109–25120. [Google Scholar] [CrossRef] [PubMed]
- Swiggers, S.J.J.; Kuijpers, M.A.; de Cort, M.J.M.; Beverloo, H.B.; Zijlmans, J.M.J.M. Critically short telomeres in acute myeloid leukemia with loss or gain of parts of chromosomes. Genes Chromosom. Cancer 2006, 45, 247–256. [Google Scholar] [CrossRef]
- Hartmann, U.; Brümmendorf, T.H.; Balabanov, S.; Thiede, C.; Illme, T.; Schaich, M. Telomere length and hTERT expression in patients with acute myeloid leukemia correlates with chromosomal abnormalities. Haematologica 2005, 90, 307–316. [Google Scholar]
- Capraro, V.; Zane, L.; Poncet, D.; Perol, D.; Galia, P.; Preudhomme, C.; Bonnefoy-Berard, N.; Gilson, E.; Thomas, X.; El-Hamri, M. Telomere deregulations possess cytogenetic, phenotype, and prognostic specificities in acute leukemias. Exp. Hematol. 2011, 39, 195–202.e2. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in dyskeratosis congenita. Blood 2009, 113, 6549–6557. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Adebayo Awe, J.; Rodrigues, P.; Kumar, R.; Houston, D.S.; Klewes, L.; Dieye, T.N.; Rego, E.M.; Passetto, R.F.; de Oliveira, F.M.; et al. Profiling Three-Dimensional Nuclear Telomeric Architecture of Myelodysplastic Syndromes and Acute Myeloid Leukemia Defines Patient Subgroups. Clin. Cancer Res. 2012, 18, 3293–3304. [Google Scholar] [CrossRef]
- Sieglová, Z.; Žilovcová, S.; Čermák, J.; Říhová, H.; Březinová, D.; Dvořáková, R.; Marková, M.; Maaloufová, J.; Sajdová, J.; Březinová, J.; et al. Dynamics of telomere erosion and its association with genome instability in myelodysplastic syndromes (MDS) and acute myelogenous leukemia arising from MDS: A marker of disease prognosis? Leuk. Res. 2004, 28, 1013–1021. [Google Scholar] [CrossRef]
- Warny, M.; Helby, J.; Sengeløv, H.; Nordestgaard, B.G.; Birgens, H.; Bojesen, S.E. Bone marrow mononuclear cell telomere length in acute myeloid leukaemia and high-risk myelodysplastic syndrome. Eur. J. Haematol. 2019, 102, 218–226. [Google Scholar] [CrossRef] [PubMed]
- XU, D.; Gruber, A.; Peterson, C.; Pisa, P. Telomerase activity and the expression of telomerase components in acute myelogenous leukaemia. Br. J. Haematol. 1998, 102, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; de Lange, T. Control of telomere length by the human telomeric protein TRF1. Nature 1997, 385, 740–743. [Google Scholar] [CrossRef]
- Vajen, B.; Thomay, K.; Schlegelberger, B. Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia. Cancers 2013, 5, 857–874. [Google Scholar] [CrossRef]
- Mai, S.; Garini, Y. The significance of telomeric aggregates in the interphase nuclei of tumor cells. J. Cell. Biochem. 2006, 97, 904–915. [Google Scholar] [CrossRef]
- Calado, R.T.; Cooper, J.N.; Padilla-Nash, H.M.; Sloand, E.M.; Wu, C.O.; Scheinberg, P.; Ried, T.; Young, N.S. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia 2012, 26, 700–707. [Google Scholar] [CrossRef]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.Y.; Moshir, S.; Mougey, V.; et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Pedersen-Bjergaard, J. Increased frequency of dicentric chromosomes in therapy-related MDS and AML compared to de novo disease is significantly related to previous treatment with alkylating agents and suggests a specific susceptibility to chromosome breakage at the centromere. Leukemia 2000, 14, 105–111. [Google Scholar] [CrossRef] [PubMed][Green Version]
- MacKinnon, R.N.; Campbell, L.J. The Role of Dicentric Chromosome Formation and Secondary Centromere Deletion in the Evolution of Myeloid Malignancy. Genet. Res. Int. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sarova, I.; Brezinova, J.; Zemanova, Z.; Ransdorfova, S.; Izakova, S.; Svobodova, K.; Pavlistova, L.; Berkova, A.; Cermak, J.; Jonasova, A.; et al. Molecular cytogenetic analysis of dicentric chromosomes in acute myeloid leukemia. Leuk. Res. 2016, 43, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Cheeseman, I.M. Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosom. Res. 2013, 21, 407–418. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Bochtler, T.; Granzow, M.; Stölzel, F.; Kunz, C.; Mohr, B.; Kartal-Kaess, M.; Hinderhofer, K.; Heilig, C.E.; Kramer, M.; Thiede, C.; et al. Marker chromosomes can arise from chromothripsis and predict adverse prognosis in acute myeloid leukemia. Blood 2017, 129, 1333–1342. [Google Scholar] [CrossRef]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rorá, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef]
- Koduru, P.R.; Wilson, K.; Wen, J.; Garcia, R.; Patel, S.; Monaghan, S.A. Cytogenetic and Cytogenomic Microarray Characterization of Chromothripsis in Chromosome 8 Affecting MOZ/NCOA2 (TIF2), FGFR1, RUNX1T1, and RUNX1 in a Pediatric Acute Myeloid Leukemia. J. Pediatr. Hematol. Oncol. 2017, 39, e227–e232. [Google Scholar] [CrossRef] [PubMed]
- Righolt, C.; Mai, S. Shattered and stitched chromosomes-chromothripsis and chromoanasynthesis-manifestations of a new chromosome crisis? Genes Chromosom. Cancer 2012, 51, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Y.-H.; Mina, A.; Altman, J.K.; Kim, K.-Y.; Zhang, Y.; Lu, X.; Jennings, L.; Sukhanova, M. Unique morphologic and genetic characteristics of acute myeloid leukemia with chromothripsis: A clinicopathologic study from a single institution. Hum. Pathol. 2020, 98, 22–31. [Google Scholar] [CrossRef]
- L′Abbate, A.; Tolomeo, D.; Cifola, I.; Severgnini, M.; Turchiano, A.; Augello, B.; Squeo, G.; D′Addabbo, P.; Traversa, D.; Daniele, G.; et al. MYC-containing amplicons in acute myeloid leukemia: Genomic structures, evolution, and transcriptional consequences. Leukemia 2018, 32, 2152–2166. [Google Scholar] [CrossRef]
- Descartes, M.; Korf, B.R.; Mikhail, F.M. Chromosomes and Chromosomal Abnormalities. In Swaiman’s Pediatric Neurology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 268–276. [Google Scholar]
- Fuse, K.; Tanaka, T.; Shibasaki, Y.; Furukawa, T.; Narita, M.; Sone, H.; Masuko, M. Marker chromosome is a strong poor prognosis factor after allogeneic HSCT for adverse-risk AML patients. Eur. J. Haematol. 2020, ejh.13495. [Google Scholar] [CrossRef]
- Liu, W.-J.; Tan, X.-H.; Luo, X.-P.; Guo, B.-P.; Wei, Z.-J.; Ke, Q.; He, S.; Cen, H. Prognostic significance of Tet methylcytosine dioxygenase 2 (TET2) gene mutations in adult patients with acute myeloid leukemia: A meta-analysis. Leuk. Lymphoma 2014, 55, 2691–2698. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Wu, L.; Zhang, Q.; Yang, R.; Yang, Y.; Zhang, Z.; He, Q.; Chang, C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: Relation to adverse epigenetic alteration and poor prognostic scoring. Ann. Hematol. 2011, 90, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, B.-R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Döhner, K.; Brown, J.; Hehmann, U.; Hetzel, C.; Stewart, J.; Lowther, G.; Scholl, C.; Fröhling, S.; Cuneo, A.; Tsui, L.C.; et al. Molecular Cytogenetic Characterization of a Critical Region in Bands 7q35-q36 Commonly Deleted in Malignant Myeloid Disorders. Blood 1998, 92, 4031–4035. [Google Scholar] [CrossRef]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of heterozygosity in 7q myeloid disorders: Clinical associations and genomic pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef]
- Mrózek, K.; Eisfeld, A.-K.; Kohlschmidt, J.; Carroll, A.J.; Walker, C.J.; Nicolet, D.; Blachly, J.S.; Bill, M.; Papaioannou, D.; Wang, E.S.; et al. Complex karyotype in de novo acute myeloid leukemia: Typical and atypical subtypes differ molecularly and clinically. Leukemia 2019, 33, 1620–1634. [Google Scholar] [CrossRef]
- Wang, J.; He, N.; Wang, R.; Tian, T.; Han, F.; Zhong, C.; Zhang, C.; Hua, M.; Ji, C.; Ma, D. Analysis of TET2 and EZH2 gene functions in chromosome instability in acute myeloid leukemia. Sci. Rep. 2020, 10, 2706. [Google Scholar] [CrossRef]
- Schvartzman, J.-M.; Duijf, P.H.G.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 Is a Critical Mediator of the Chromosome Instability Observed upon Rb and p53 Pathway Inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef]
- Sotillo, R.; Hernando, E.; Díaz-Rodríguez, E.; Teruya-Feldstein, J.; Cordón-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 Overexpression Promotes Aneuploidy and Tumorigenesis in Mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef]
- Mondal, G.; Sengupta, S.; Panda, C.K.; Gollin, S.M.; Saunders, W.S.; Roychoudhury, S. Overexpression of Cdc20 leads to impairment of the spindle assembly checkpoint and aneuploidization in oral cancer. Carcinogenesis 2007, 28, 81–92. [Google Scholar] [CrossRef]
- Simonetti, G.; Padella, A.; do Valle, I.F.; Fontana, M.C.; Fonzi, E.; Bruno, S.; Baldazzi, C.; Guadagnuolo, V.; Manfrini, M.; Ferrari, A.; et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer 2019, 125, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kantarjian, H.; Wang, H.; Cortes, J.; Ravandi, F. Acute promyelocytic leukemia: A population-based study on incidence and survival in the United States, 1975–2008. Cancer 2012, 118, 5811–5818. [Google Scholar] [CrossRef]
- Labrador, J.; Luño, E.; Vellenga, E.; Brunet, S.; González-Campos, J.; Chillón, M.C.; Holowiecka, A.; Esteve, J.; Bergua, J.; González-Sanmiguel, J.D.; et al. Clinical significance of complex karyotype at diagnosis in pediatric and adult patients with de novo acute promyelocytic leukemia treated with ATRA and chemotherapy. Leuk. Lymphoma 2019, 60, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Lushnikova, T.; Bouska, A.; Odvody, J.; Dupont, W.D.; Eischen, C.M. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene 2011, 30, 4622–4631. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, C.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A. Bone Marrow Senescence and the Microenvironment of Hematological Malignancies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Vaziri, H.; Dragowska, W.; Allsopp, R.C.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA 1994, 91, 9857–9860. [Google Scholar] [CrossRef]
- Silva, P.; Neumann, M.; Schroeder, M.P.; Vosberg, S.; Schlee, C.; Isaakidis, K.; Ortiz-Tanchez, J.; Fransecky, L.R.; Hartung, T.; Türkmen, S.; et al. Acute myeloid leukemia in the elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia 2017, 31, 1640–1644. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.A.L.M.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef]
- Granfeldt Østgård, L.S.; Medeiros, B.C.; Sengeløv, H.; Nørgaard, M.; Andersen, M.K.; Dufva, I.H.; Friis, L.S.; Kjeldsen, E.; Marcher, C.W.; Preiss, B.; et al. Epidemiology and Clinical Significance of Secondary and Therapy-Related Acute Myeloid Leukemia: A National Population-Based Cohort Study. J. Clin. Oncol. 2015, 33, 3641–3649. [Google Scholar] [CrossRef]
- Kern, W.; Haferlach, T.; Schnittger, S.; Ludwig, W.; Hiddemann, W.; Schoch, C. Karyotype instability between diagnosis and relapse in 117 patients with acute myeloid leukemia: Implications for resistance against therapy. Leukemia 2002, 16, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Thomay, K.; Hofmann, W.; Buesche, G.; Kreipe, H.; Thol, F.; Heuser, M.; Ganser, A.; Schlegelberger, B.; Göhring, G. Routes of Clonal Evolution into Complex Karyotypes in Myelodysplastic Syndrome Patients with 5q Deletion. Int. J. Mol. Sci. 2018, 19, 3269. [Google Scholar] [CrossRef] [PubMed]
- Moison, C.; Lavallée, V.-P.; Thiollier, C.; Lehnertz, B.; Boivin, I.; Mayotte, N.; Gareau, Y.; Fréchette, M.; Blouin-Chagnon, V.; Corneau, S.; et al. Complex karyotype AML displays G2/M signature and hypersensitivity to PLK1 inhibition. Blood Adv. 2019, 3, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.M.K.; Zhang, C.; Ng, N.K.L.; Yang, N.; Lam, S.S.Y.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; Tsui, S.P.; Ip, H.W.; et al. Distinct mutation spectrum, clinical outcome and therapeutic responses of typical complex/monosomy karyotype acute myeloid leukemia carrying TP53 mutations. Am. J. Hematol. 2019, 94, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Ouillette, P.; Li, Y.; Keller, J.; Lam, C.; Roulston, D.; Li, C.; Shedden, K.; Malek, S.N. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood 2013, 121, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Fröhling, S.; Krämer, A. Role of chromosomal aberrations in clonal diversity and progression of acute myeloid leukemia. Leukemia 2015, 29, 1243–1252. [Google Scholar] [CrossRef]
- Hou, H.-A.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Lin, L.-I.; Tseng, M.-H.; Chiang, Y.-C.; Liu, M.-C.; Liu, C.-W.; Tang, J.-L.; et al. TP53 mutations in de novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Walter-petrich, A.; Lehmann che, J.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. APR-246 Combined with Azacitidine (AZA) in TP53 Mutated Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). a Phase 2 Study By the Groupe Francophone Des Myélodysplasies (GFM). Blood 2019, 134, 677. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.F.; McGraw, K.; et al. Phase 2 Results of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2019, 134, 676. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53 -Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Göhring, G.; Giagounidis, A.; Büsche, G.; Kreipe, H.H.; Zimmermann, M.; Hellström-Lindberg, E.; Aul, C.; Schlegelberger, B. Patients with del(5q) MDS who fail to achieve sustained erythroid or cytogenetic remission after treatment with lenalidomide have an increased risk for clonal evolution and AML progression. Ann. Hematol. 2010, 89, 365–374. [Google Scholar] [CrossRef]
- Middeke, J.M.; Beelen, D.; Stadler, M.; Göhring, G.; Schlegelberger, B.; Baurmann, H.; Bug, G.; Bellos, F.; Mohr, B.; Buchholz, S.; et al. Outcome of high-risk acute myeloid leukemia after allogeneic hematopoietic cell transplantation: Negative impact of abnl(17p) and −5/5q−. Blood 2012, 120, 2521–2528. [Google Scholar] [CrossRef]
- Bulaeva, E.; Pellacani, D.; Nakamichi, N.; Hammond, C.A.; Beer, P.A.; Lorzadeh, A.; Moksa, M.; Carles, A.; Bilenky, M.; Lefort, S.; et al. MYC-induced human acute myeloid leukemia requires a continuing IL-3/GM-CSF costimulus. Blood 2020, 136, 2764–2773. [Google Scholar] [CrossRef]
- Pan, X.-N.; Chen, J.-J.; Wang, L.-X.; Xiao, R.-Z.; Liu, L.-L.; Fang, Z.-G.; Liu, Q.; Long, Z.-J.; Lin, D.-J. Inhibition of c-Myc Overcomes Cytotoxic Drug Resistance in Acute Myeloid Leukemia Cells by Promoting Differentiation. PLoS ONE 2014, 9, e105381. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Dawson, M.; Stein, E.M.; Huntly, B.J.P.; Karadimitris, A.; Kamdar, M.; Fernandez de Larrea, C.; Dickinson, M.J.; Yeh, P.S.-H.; Daver, N.; Chaidos, A.; et al. A Phase I Study of GSK525762, a Selective Bromodomain (BRD) and Extra Terminal Protein (BET) Inhibitor: Results from Part 1 of Phase I/II Open Label Single Agent Study in Patients with Acute Myeloid Leukemia (AML). Blood 2017, 130, 1377. [Google Scholar] [CrossRef]
- Kang, C.; Kim, C.-Y.; Kim, H.S.; Park, S.-P.; Chung, H.-M. The Bromodomain Inhibitor JQ1 Enhances the Responses to All- trans Retinoic Acid in HL-60 and MV4-11 Leukemia Cells. Int. J. Stem Cells 2018, 11, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Call, S.G.; Duren, R.P.; Panigrahi, A.K.; Nguyen, L.; Freire, P.R.; Grimm, S.L.; Coarfa, C.; Conneely, O.M. Targeting Oncogenic Super Enhancers in MYC-Dependent AML Using a Small Molecule Activator of NR4A Nuclear Receptors. Sci. Rep. 2020, 10, 2851. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Blonquist, T.M.; DeAngelo, D.J.; McMasters, M.; Winer, E.S.; Hobbs, G.S.; Amrein, P.C.; Hock, H.; Steensma, D.P.; Garcia, J.S.; et al. Phase II Clinical Trial of Alisertib, an Aurora a Kinase Inhibitor, in Combination with Induction Chemotherapy in High-Risk, Untreated Patients with Acute Myeloid Leukemia. Blood 2018, 132, 766. [Google Scholar] [CrossRef]
- Sarova, I.; Brezinova, J.; Zemanova, Z.; Ransdorfova, S.; Svobodova, K.; Izakova, S.; Pavlistova, L.; Lizcova, L.; Berkova, A.; Skipalova, K.; et al. High frequency of dicentric chromosomes detected by multi-centromeric FISH in patients with acute myeloid leukemia and complex karyotype. Leuk. Res. 2018, 68, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.L.; Fenaux, P.; Craig, M.D.; Gyan, E.; Lister, J.; Kassis, J.; Pigneux, A.; Schiller, G.J.; Jung, J.; Jane Leonard, E.; et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk. Res. Rep. 2014, 3, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Wander, S.A.; Blonquist, T.M.; Brunner, A.M.; Amrein, P.C.; Supko, J.; Hermance, N.M.; Manning, A.L.; Sadrzadeh, H.; Ballen, K.K.; et al. Phase I study of the aurora A kinase inhibitor alisertib with induction chemotherapy in patients with acute myeloid leukemia. Haematologica 2017, 102, 719–727. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Payton, M.; Cheung, H.-K.; Ninniri, M.S.S.; Marinaccio, C.; Wayne, W.C.; Hanestad, K.; Crispino, J.D.; Juan, G.; Coxon, A. Dual Targeting of Aurora Kinases with AMG 900 Exhibits Potent Preclinical Activity Against Acute Myeloid Leukemia with Distinct Post-Mitotic Outcomes. Mol. Cancer Ther. 2018, 17, 2575–2585. [Google Scholar] [CrossRef]
- Qi, J.; Gao, X.; Zhong, X.; Zhang, N.; Wang, R.; Zhang, H.; Pan, T.; Liu, X.; Yao, Y.; Wu, Q.; et al. Selective inhibition of Aurora A and B kinases effectively induces cell cycle arrest in t(8;21) acute myeloid leukemia. Biomed. Pharmacother. 2019, 117, 109113. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.; Davies, M.; Oliver, S.; D’Souza, R.; Pike, L.; Stockman, P. Phase I study of the Aurora B kinase inhibitor barasertib (AZD1152) to assess the pharmacokinetics, metabolism and excretion in patients with acute myeloid leukemia. Cancer Chemother. Pharmacol. 2012, 70, 461–469. [Google Scholar] [CrossRef]
- Löwenberg, B.; Muus, P.; Ossenkoppele, G.; Rousselot, P.; Cahn, J.-Y.; Ifrah, N.; Martinelli, G.; Amadori, S.; Berman, E.; Sonneveld, P.; et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 2011, 118, 6030–6036. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Yokozawa, T.; Sakura, T.; Watanabe, T.; Fujisawa, S.; Yamauchi, T.; Uike, N.; Ando, K.; Kihara, R.; Tobinai, K.; et al. A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk. Res. 2011, 35, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Sekeres, M.A.; Ribrag, V.; Rousselot, P.; Garcia-Manero, G.; Jabbour, E.J.; Owen, K.; Stockman, P.K.; Oliver, S.D. Phase I Study Assessing the Safety and Tolerability of Barasertib (AZD1152) With Low-Dose Cytosine Arabinoside in Elderly Patients With AML. Clin. Lymphoma Myeloma Leuk. 2013, 13, 559–567. [Google Scholar] [CrossRef]
- Hartsink-Segers, S.A.; Zwaan, C.M.; Exalto, C.; Luijendijk, M.W.J.; Calvert, V.S.; Petricoin, E.F.; Evans, W.E.; Reinhardt, D.; de Haas, V.; Hedtjärn, M.; et al. Aurora kinases in childhood acute leukemia: The promise of aurora B as therapeutic target. Leukemia 2013, 27, 560–568. [Google Scholar] [CrossRef]
- Ikezoe, T.; Yang, J.; Nishioka, C.; Yokoyama, A. p53 is critical for the Aurora B kinase inhibitor-mediated apoptosis in acute myelogenous leukemia cells. Int. J. Hematol. 2009, 91, 69–77. [Google Scholar] [CrossRef]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.-H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Chou, S.-C.; Liu, C.-Y.; Chen, C.-Y.; Hou, H.-A.; Kuo, Y.-Y.; Lee, M.-C.; Ko, B.-S.; Tang, J.-L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [PubMed]
- Das, A.B.; Kakadia, P.M.; Wojcik, D.; Pemberton, L.; Browett, P.J.; Bohlander, S.K.; Vissers, M.C.M. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J. 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Kayser, S.; Döhner, K.; Krauter, J.; Köhne, C.-H.; Horst, H.A.; Held, G.; von Lilienfeld-Toal, M.; Wilhelm, S.; Kündgen, A.; Götze, K.; et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145. [Google Scholar] [CrossRef]
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar] [CrossRef]
- Byrd, J.; Mrózek, K.; Dodge, R.; Carroll, A.; Edwards, C.; Arthur, D.; Pettenati, M.; Patil, S.; Rao, K.; Watson, M.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Schoch, C.; Kern, W.; Kohlmann, A.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosom. Cancer 2005, 43, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.M.; Dumitriu, B.; Hilden, P.; Chen, C.; Rapaport, F.; Kishtagari, A.; Ahn, J.; Devlin, S.M.; Rampal, R.K.; Levine, R.L.; et al. Telomere Length Is Associated with Specific Mutations and Mutation Classes in Patients with Acute Myeloid Leukemia. Blood 2014, 124, 2280. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Koh, Y.; Kim, I.; Bae, J.-Y.; Song, E.Y.; Kim, H.-K.; Yoon, S.-S.; Lee, D.S.; Park, S.S.; Park, M.H.; Park, S.; et al. Prognosis of Secondary Acute Myeloid Leukemia is Affected by the Type of the Preceding Hematologic Disorders and the Presence of Trisomy 8. Jpn. J. Clin. Oncol. 2010, 40, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dai, H.; Wang, Q.; Wang, Q.; Xu, Y.; Wang, Y.; Sun, A.; Ruan, J.; Chen, S.; Wu, D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. PLoS ONE 2013, 8, e61341. [Google Scholar] [CrossRef][Green Version]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: An analysis in 1604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Nawrocki, S.T.; Espitia, C.M.; Zhang, M.; Yang, J.J.; Padmanabhan, S.; Ecsedy, J.; Giles, F.J.; Carew, J.S. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int. J. Cancer 2012, 131, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, C.; Johnson, V.L.; Tighe, A.; Ellston, R.; Haworth, C.; Johnson, T.; Mortlock, A.; Keen, N.; Taylor, S.S. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 2003, 161, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Goldenson, B.; Silver, S.J.; Schenone, M.; Dancik, V.; Huang, Z.; Wang, L.-Z.; Lewis, T.A.; An, W.F.; Li, X.; et al. Identification of Regulators of Polyploidization Presents Therapeutic Targets for Treatment of AMKL. Cell 2012, 150, 575–589. [Google Scholar] [CrossRef]
- Krause, D.S.; Crispino, J.D. Molecular Pathways: Induction of Polyploidy as a Novel Differentiation Therapy for Leukemia. Clin. Cancer Res. 2013, 19, 6084–6088. [Google Scholar] [CrossRef]
- Bruedigam, C.; Bagger, F.O.; Heidel, F.H.; Paine Kuhn, C.; Guignes, S.; Song, A.; Austin, R.; Vu, T.; Lee, E.; Riyat, S.; et al. Telomerase Inhibition Effectively Targets Mouse and Human AML Stem Cells and Delays Relapse following Chemotherapy. Cell Stem Cell 2014, 15, 775–790. [Google Scholar] [CrossRef]
- Hidaka, D.; Onozawa, M.; Miyashita, N.; Yokoyama, S.; Nakagawa, M.; Hashimoto, D.; Teshima, T. Short-term treatment with imetelstat sensitizes hematopoietic malignant cells to a genotoxic agent via suppression of the telomerase-mediated DNA repair process. Leuk. Lymphoma 2020, 61, 2722–2732. [Google Scholar] [CrossRef]
- Rusbuldt, J.J.; Luistro, L.; Chin, D.; Smith, M.; Wong, A.; Romero, M.; Rizo, A.; Bussolari, J.; Huang, F.; Sasser, A. (Kate) Abstract 1101: Telomerase inhibitor imetelstat in combination with the BCL-2 inhibitor venetoclax enhances apoptosis in vitro and increases survival in vivo in acute myeloid leukemia. In Proceedings of the Experimental and Molecular Therapeutics, Washington, DC, USA, 1–5 April 2017; American Association for Cancer Research: Philadelphia, PA, USA, 2017; p. 1101. [Google Scholar]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mingay, M.; Chaturvedi, A.; Bilenky, M.; Cao, Q.; Jackson, L.; Hui, T.; Moksa, M.; Heravi-Moussavi, A.; Humphries, R.K.; Heuser, M.; et al. Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia 2018, 32, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Renner, A.G.; Dos Santos, C.; Recher, C.; Bailly, C.; Créancier, L.; Kruczynski, A.; Payrastre, B.; Manenti, S. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009, 114, 659–662. [Google Scholar] [CrossRef]
- Cunningham, C.E.; MacAuley, M.J.; Vizeacoumar, F.S.; Abuhussein, O.; Freywald, A.; Vizeacoumar, F.J. The CINs of Polo-Like Kinase 1 in Cancer. Cancers 2020, 12, 2953. [Google Scholar] [CrossRef]
- Ikezoe, T.; Yang, J.; Nishioka, C.; Takezaki, Y.; Tasaka, T.; Togitani, K.; Koeffler, H.P.; Yokoyama, A. A novel treatment strategy targeting polo-like kinase 1 in hematological malignancies. Leukemia 2009, 23, 1564–1576. [Google Scholar] [CrossRef]
- Brandwein, J.M. Targeting polo-like kinase 1 in acute myeloid leukemia. Ther. Adv. Hematol. 2015, 6, 80–87. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Berneman, Z.N.; Anguille, S.; Van Marck, V.; Schroyens, W.A.; Van Tendeloo, V.F. Induction of complete remission of acute myeloid leukaemia by pegylated interferon-α-2a in a patient with transformed primary myelofibrosis. Br. J. Haematol. 2010, 149, 152–155. [Google Scholar] [CrossRef]
- Anguille, S.; Lion, E.; Willemen, Y.; Van Tendeloo, V.F.I.; Berneman, Z.N.; Smits, E.L.J.M. Interferon-α in acute myeloid leukemia: An old drug revisited. Leukemia 2011, 25, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.J.M.; Anguille, S.; Berneman, Z.N. Interferon α may be back on track to treat acute myeloid leukemia. Oncoimmunology 2013, 2, e23619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical Relationship between Chromosomal Instability and Survival Outcome in Cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of Paclitaxel in Breast Cancer Is due to Chromosome Missegregation on Multipolar Spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; A’Hern, R.; Birkbak, N.J.; Gorman, P.; Grönroos, E.; Ngang, S.; Nicola, P.; Rahman, L.; Thanopoulou, E.; Kelly, G.; et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: A prospective validation cohort study from the TACT trial. Ann. Oncol. 2015, 26, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N. The Role of Inhibition of Apoptosis in Acute Leukemias and Myelodysplastic Syndrome. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e6. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef]
- Esposito, M.T.; So, C.W.E. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.-S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; Eric So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 462–472. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Features | Promising Agents 1 | |
|---|---|---|
| CK | [20,186,187] | PLK1 inhibitors [188]. In TP53 mutated: APR-246 [189] |
| Aneuploidy | [190,191] | PLK1 inhibitors [188]. In TP53 mutated: APR-246 [189] |
| TP53 | [192,193,194] | APR-246 with azacitidine (AZA) [195,196]. APR-246 alone [189], APR-246 combined with azacitidine [197,198]. |
| Abnormal 5q | [64,187,199,200] | PLK1 inhibitors [188]. In TP53 mutated: APR-246 [189] APR-246 with azacitidine [195,196]. APR-246 alone [189] |
| MYC abnormalities | [201,202] | Molibresib (GSK525762) [203,204] JQ1 combined with All-trans retinoic acid (ATRA) [205] Dihydroergotamine (DHE) [206] |
| Trisomy 8 | [118,119,120,121] | Molibresib (GSK525762) [203,204] JQ1 combined with All-trans retinoic acid (ATRA) [205] Dihydroergotamine (DHE) [206] |
| Telomere dysfunction | [136,144] | (hTERT) JQ1 combined with All-trans retinoic acid (ATRA) [205] Telomerase inhibitors [201,207]. |
| Dicentric chromosomes (DCs) | [150,208] | PLK1 inhibitors [188]. In TP53 mutated: APR-246 [189] APR-246 with azacitidine [195,196]. APR-246 alone [189] |
| Aurora Kinases | [100,101,102,103,209] | Small molecule inhibitors [210,211,212,213,214,215,216,217,218,219,220] |
| TET2 abnormalities | [173,221,222] | Decitabine [173]. Vitamin C combined with PARP inhibitor [223]. Ascorbate (Vitamin C) [224] |
| EZH2 abnormalities | [173,225] | Decitabine [173] |
| Features | de novo AML | sAML | tAML | References |
|---|---|---|---|---|
| Chromosomal abnormalities | 47–51% | 62.2% | 75–92% | [185,226,227] |
| Adverse cytogenetics | 19% | 22% | 39–40% | [185,226] |
| Complex karyotype (CK) | 10–23% | 18% | 26- 40% | [24,25,226] |
| Abnormal 17p | 5–11% | 14% | 14% | [33,226,228] |
| TP53 Mutation | 7–21% | 15% | 47% | [192,229] |
| −5 or 5q− | 5–8% | 7–14% | 14–21% | [25,33,226,227] |
| Abnormal chromosome 7 | 2–7% | 19% | 10% | [25,33,226,230] |
| Telomere Length: TRF | No significant differences | ND | [141,231] | |
| MYC overexpression | 35% | 27% | ND | [232] |
| +8 | 10–25% | 25% | 26% | [228,233] |
| Dicentric Chromosomes (DCs) | 3% | ND | 15% | [149] |
| DCs in CK cohorts | 48% | 59% | ND | [208] |
| TET2 mutations | 13–27% | 24–29% | 24% | [222,234,235] |
| EZH2 abnormalities | 2–4% | 7–9% | 8% | [229,236,237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira Lisboa, M.; Brofman, P.R.S.; Schmid-Braz, A.T.; Rangel-Pozzo, A.; Mai, S. Chromosomal Instability in Acute Myeloid Leukemia. Cancers 2021, 13, 2655. https://doi.org/10.3390/cancers13112655
de Oliveira Lisboa M, Brofman PRS, Schmid-Braz AT, Rangel-Pozzo A, Mai S. Chromosomal Instability in Acute Myeloid Leukemia. Cancers. 2021; 13(11):2655. https://doi.org/10.3390/cancers13112655
Chicago/Turabian Stylede Oliveira Lisboa, Mateus, Paulo Roberto Slud Brofman, Ana Teresa Schmid-Braz, Aline Rangel-Pozzo, and Sabine Mai. 2021. "Chromosomal Instability in Acute Myeloid Leukemia" Cancers 13, no. 11: 2655. https://doi.org/10.3390/cancers13112655
APA Stylede Oliveira Lisboa, M., Brofman, P. R. S., Schmid-Braz, A. T., Rangel-Pozzo, A., & Mai, S. (2021). Chromosomal Instability in Acute Myeloid Leukemia. Cancers, 13(11), 2655. https://doi.org/10.3390/cancers13112655