
Biomarkers in Hepatobiliary Cancers: What Is Useful in Clinical Practice?

 ,
, {kind=link}
{kind=link}
{kind=link}
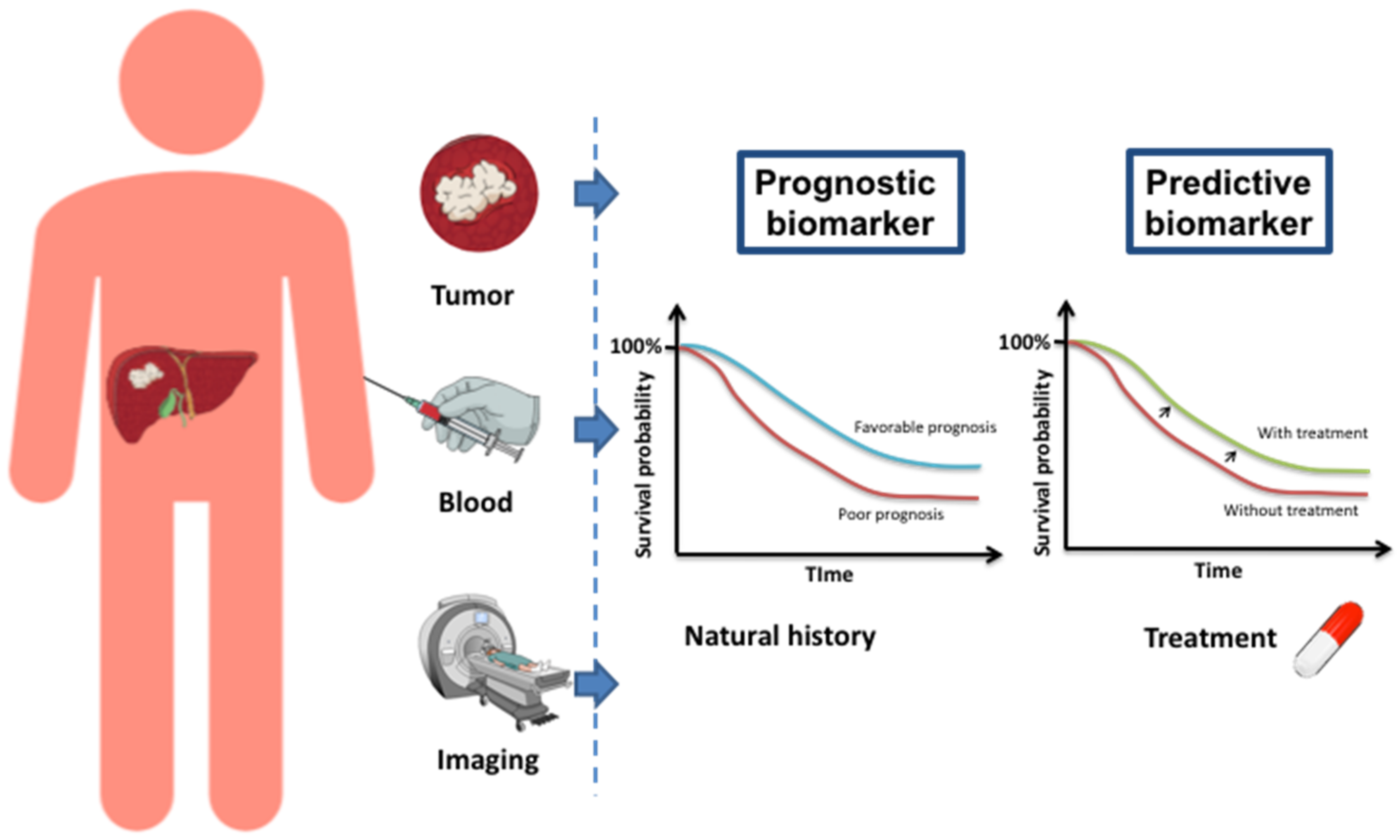
Abstract
:Simple Summary
Abstract
1. Introduction
2. Main Prognostic Biomarkers in Hepatocellular Carcinoma and in Biliary Tract Cancer
2.1. In Hepatocellular Carcinoma
2.1.1. Classical Biomarkers
2.1.2. Emerging Biomarkers
2.2. In Biliary Tract Cancers
2.2.1. Classical Biomarkers
2.2.2. Emerging Biomarkers
Molecular Biomarkers
Immune Microenvironment
2.2.3. Circulating Tumor Cells
3. Predictive Markers of Treatment Response
3.1. In Hepatocellular Carcinoma
3.1.1. Angiogenesis Inhibitors
3.1.2. Immunotherapies
3.2. In Biliary Tract Cancers
3.2.1. Targeted Therapies
3.2.2. Immunotherapies
4. What is Useful in Clinical Practice?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.-M. Pathophysiology of Hepatitis C Virus Infection and Related Liver Disease. Trends Microbiol. 2004, 12, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B Virus Infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-Dependent Mechanisms of Hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Vanni, E.; Marchesini, G. NASH and the Risk of Cirrhosis and Hepatocellular Carcinoma in Type 2 Diabetes. Curr. Diab. Rep. 2007, 7, 175–180. [Google Scholar] [CrossRef]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and Hepatocellular Carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Classification, Diagnosis, and Management of Cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in Pathogenesis, Diagnosis, and Treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Patel, T. Cholangiocarcinoma–Controversies and Challenges. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert Consensus Document: Cholangiocarcinoma: Current Knowledge and Future Perspectives Consensus Statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Charbel, H.; Al-Kawas, F.H. Cholangiocarcinoma: Epidemiology, Risk Factors, Pathogenesis, and Diagnosis. Curr. Gastroenterol. Rep. 2011, 13, 182–187. [Google Scholar] [CrossRef]
- Hennedige, T.P.; Neo, W.T.; Venkatesh, S.K. Imaging of Malignancies of the Biliary Tract- an Update. Cancer Imaging 2014, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Patel, T. Worldwide Trends in Mortality from Biliary Tract Malignancies. BMC Cancer 2002, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Benipal, B. Incidence of Cholangiocarcinoma in the USA from 2001 to 2015: A US Cancer Statistics Analysis of 50 States. Cureus 2001, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirstein, M.M.; Vogel, A. Epidemiology and Risk Factors of Cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19. [Google Scholar] [CrossRef]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk Factors for Intrahepatic and Extrahepatic Cholangiocarcinoma: A Systematic Review and Meta-Analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular Carcinoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Dondorf, F.; Uteβ, F.; Fahrner, R.; Felgendreff, P.; Ardelt, M.; Tautenhahn, H.-M.; Settmacher, U.; Rauchfuβ, F. Liver Transplant for Perihilar Cholangiocarcinoma (Klatskin Tumor): The Essential Role of Patient Selection. Exp. Clin. Transplant. 2019, 17, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Friman, S. Cholangiocarcinoma–Current Treatment Options. Scand. J. Surg. 2011, 100, 30–34. [Google Scholar] [CrossRef]
- Wu, L.; Tsilimigras, D.I.; Farooq, A.; Hyer, J.M.; Merath, K.; Paredes, A.Z.; Mehta, R.; Sahara, K.; Shen, F.; Pawlik, T.M. Potential Survival Benefit of Radiofrequency Ablation for Small Solitary Intrahepatic Cholangiocarcinoma in Nonsurgically Managed Patients: A Population-Based Analysis. J. Surg. Oncol. 2019, 120, 1358–1364. [Google Scholar] [CrossRef]
- Mavros, M.N.; Economopoulos, K.P.; Alexiou, V.G.; Pawlik, T.M. Treatment and Prognosis for Patients with Intrahepatic Cholangiocarcinoma: Systematic Review and Meta-Analysis. JAMA Surg. 2014, 149, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.G.; Song, G.W.; Hwang, S.; Ha, T.Y.; Moon, D.B.; Jung, D.H.; Kim, K.H.; Ahn, C.S.; Kim, M.H.; Lee, S.K.; et al. Surgical Treatment of Hilar Cholangiocarcinoma in the New Era: The Asan Experience. J. Hepatobiliary Pancreat. Sci. 2010, 17, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Nagino, M.; Ebata, T.; Yokoyama, Y.; Igami, T.; Sugawara, G.; Takahashi, Y.; Nimura, Y. Evolution of Surgical Treatment for Perihilar Cholangiocarcinoma: A Single-Center 34-Year Review of 574 Consecutive Resections. Ann. Surg. 2013, 258, 129–140. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Garcea, G.; Dennison, A.R.; Maddern, G.J. The Surgical Management of Klatskin Tumours: Has Anything Changed in the Last Decade? World J. Surg. 2015, 39, 2748–2756. [Google Scholar] [CrossRef]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D.; ESMO Guidelines Committee. Biliary Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-Line FOLFOX Chemotherapy versus Active Symptom Control for Advanced Biliary Tract Cancer (ABC-06): A Phase 3, Open-Label, Randomised, Controlled Trial. Lancet Oncol. 2021. [Google Scholar] [CrossRef]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision Medicine for Patients with Advanced Biliary Tract Cancers: An Effective Strategy within the Prospective MOSCATO-01 Trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-Mutant, Chemotherapy-Refractory Cholangiocarcinoma (ClarIDHy): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and Other Tools) Resource; Food and Drug Administration: Silver Spring, MD, USA, 2016.
- European Association for the Study of the Liver. European Association for the Study of the Liver EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Peña, C.E.A.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J.; SHARP Investigators Study Group. Plasma Biomarkers as Predictors of Outcome in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñero, F.; Dirchwolf, M.; Pessôa, M.G. Biomarkers in Hepatocellular Carcinoma: Diagnosis, Prognosis and Treatment Response Assessment. Cells 2020, 9, 1370. [Google Scholar] [CrossRef]
- Bruix, J.; Cheng, A.-L.; Meinhardt, G.; Nakajima, K.; de Sanctis, Y.; Llovet, J. Prognostic Factors and Predictors of Sorafenib Benefit in Patients with Hepatocellular Carcinoma: Analysis of Two Phase III Studies. J. Hepatol. 2017, 67, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Casadei Gardini, A.; Scarpi, E.; Faloppi, L.; Scartozzi, M.; Silvestris, N.; Santini, D.; de Stefano, G.; Marisi, G.; Negri, F.V.; Foschi, F.G.; et al. Immune Inflammation Indicators and Implication for Immune Modulation Strategies in Advanced Hepatocellular Carcinoma Patients Receiving Sorafenib. Oncotarget 2016, 7, 67142–67149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, J.; Pinato, D.J.; Ramaswami, R.; Arizumi, T.; Ferrari, C.; Gibbin, A.; Burlone, M.E.; Guaschino, G.; Toniutto, P.; Black, J.; et al. Integration of the Cancer-Related Inflammatory Response as a Stratifying Biomarker of Survival in Hepatocellular Carcinoma Treated with Sorafenib. Oncotarget 2017, 8, 36161–36170. [Google Scholar] [CrossRef]
- da Fonseca, L.G.; Barroso-Sousa, R.; Bento, A.d.S.A.; Blanco, B.P.; Valente, G.L.; Pfiffer, T.E.F.; Hoff, P.M.; Sabbaga, J. Pre-Treatment Neutrophil-to-Lymphocyte Ratio Affects Survival in Patients with Advanced Hepatocellular Carcinoma Treated with Sorafenib. Med. Oncol 2014, 31, 264. [Google Scholar] [CrossRef]
- Kinoshita, A.; Onoda, H.; Imai, N.; Iwaku, A.; Oishi, M.; Tanaka, K.; Fushiya, N.; Koike, K.; Nishino, H.; Matsushima, M.; et al. The Glasgow Prognostic Score, an Inflammation Based Prognostic Score, Predicts Survival in Patients with Hepatocellular Carcinoma. BMC Cancer 2013, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Hilmi, M.; Jouinot, A.; Burns, R.; Pigneur, F.; Mounier, R.; Gondin, J.; Neuzillet, C.; Goldwasser, F. Body Composition and Sarcopenia: The next-Generation of Personalized Oncology and Pharmacology? Pharmacol. Ther. 2019, 196, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Tselikas, L.; Pietrasz, D.; Pigneur, F.; Laurent, A.; Compagnon, P.; Salloum, C.; Luciani, A.; Azoulay, D. Sarcopenia Impacts on Short- and Long-Term Results of Hepatectomy for Hepatocellular Carcinoma. Ann. Surg. 2015, 261, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, S.; Mizuta, T.; Otsuka, T.; Nakashita, S.; Ide, Y.; Miyoshi, A.; Kitahara, K.; Eguchi, Y.; Ozaki, I.; Anzai, K. Sarcopenia is a Risk Factor for the Recurrence of Hepatocellular Carcinoma after Curative Treatment. Hepatol. Res. 2016, 46, 201–208. [Google Scholar] [CrossRef]
- Meza-Junco, J.; Montano-Loza, A.J.; Baracos, V.E.; Prado, C.M.M.; Bain, V.G.; Beaumont, C.; Esfandiari, N.; Lieffers, J.R.; Sawyer, M.B. Sarcopenia as a Prognostic Index of Nutritional Status in Concurrent Cirrhosis and Hepatocellular Carcinoma. J. Clin. Gastroenterol. 2013, 47, 861–870. [Google Scholar] [CrossRef]
- Levolger, S.; van Vledder, M.G.; Muslem, R.; Koek, M.; Niessen, W.J.; de Man, R.A.; de Bruin, R.W.F.; Ijzermans, J.N.M. Sarcopenia Impairs Survival in Patients with Potentially Curable Hepatocellular Carcinoma. J. Surg. Oncol. 2015, 112, 208–213. [Google Scholar] [CrossRef]
- Mir, O.; Coriat, R.; Boudou-Rouquette, P.; Ropert, S.; Durand, J.-P.; Cessot, A.; Mallet, V.; Sogni, P.; Chaussade, S.; Pol, S.; et al. Gemcitabine and Oxaliplatin as Second-Line Treatment in Patients with Hepatocellular Carcinoma Pre-Treated with Sorafenib. Med. Oncol. 2012, 29, 2793–2799. [Google Scholar] [CrossRef]
- Sachdeva, M.; Arora, S.K. Prognostic Role of Immune Cells in Hepatocellular Carcinoma. EXCLI J. 2020, 19, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of Immune Microenvironment in Hepatocellular Carcinoma and Its Additional Impact on Histological and Molecular Classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lou, J.; Fu, L.; Jin, Q. An Independent Poor-Prognosis Subtype of Hepatocellular Carcinoma Based on the Tumor Microenvironment. J. Int. Med. Res. 2021, 49. [Google Scholar] [CrossRef]
- Chen, W.; Tang, D.; Ou, M.; Dai, Y. Mining Prognostic Biomarkers of Hepatocellular Carcinoma Based on Immune-Associated Genes. DNA Cell Biol. 2020, 39, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Zhang, Y. Integrated Analysis of Immunity- and Ferroptosis-Related Biomarker Signatures to Improve the Prognosis Prediction of Hepatocellular Carcinoma. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, Y.; Okano, S.; Matsumoto, Y.; Nakagawara, H.; Matono, R.; Yoshiya, S.; Yamashita, Y.-I.; Yoshizumi, T.; Ikegami, T.; Soejima, Y.; et al. Prognostic Impact of Programmed Cell Death 1 Ligand 1 Expression in Human Leukocyte Antigen Class I-Positive Hepatocellular Carcinoma after Curative Hepatectomy. J. Gastroenterol. 2015, 50, 65–75. [Google Scholar] [CrossRef]
- Jung, H.I.; Jeong, D.; Ji, S.; Ahn, T.S.; Bae, S.H.; Chin, S.; Chung, J.C.; Kim, H.C.; Lee, M.S.; Baek, M.-J. Overexpression of PD-L1 and PD-L2 is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Cancer Res. Treat. 2017, 49, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed Death Ligand 1 Expression in Hepatocellular Carcinoma: Relationship with Clinical and Pathological Features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.-W.; An, L.; Huang, H.-Y.; Sun, X.-D.; Lv, G.-Y. Establishing Peripheral PD-L1 as a Prognostic Marker in Hepatocellular Carcinoma Patients: How Long Will It Come True? Clin. Transl. Oncol. 2021, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome Classification of HCC is Related to Gene Alterations and to New Therapeutic Targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and Histological Correlations in Liver Cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.-F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological Subtypes of Hepatocellular Carcinoma are Related to Gene Mutations and Molecular Tumour Classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Zhang, S.; Chang, L. Expression of Cell Divisioncycle-Associated Genes and Their Prognostic Significance in Hepatocellular Carcinoma. Int. J. Clin. Exp. Pathol. 2021, 14, 151–169. [Google Scholar]
- Trevisan França de Lima, L.; Broszczak, D.; Zhang, X.; Bridle, K.; Crawford, D.; Punyadeera, C. The Use of Minimally Invasive Biomarkers for the Diagnosis and Prognosis of Hepatocellular Carcinoma. Biochim. Biophys. Acta Rev. Cancer 2020, 1874. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.C.; Teng, P.-C.; Chen, P.-J.; Posadas, E.; Tseng, H.-R.; Lu, S.C.; Yang, J.D. Detection of Circulating Tumor Cells and Their Implications as a Biomarker for Diagnosis, Prognostication, and Therapeutic Monitoring in Hepatocellular Carcinoma. Hepatology 2021, 73, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Mjelle, R.; Dima, S.O.; Bacalbasa, N.; Chawla, K.; Sorop, A.; Cucu, D.; Herlea, V.; Sætrom, P.; Popescu, I. Comprehensive Transcriptomic Analyses of Tissue, Serum, and Serum Exosomes from Hepatocellular Carcinoma Patients. BMC Cancer 2019, 19, 1007. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to Build a Bridge from a Population-Based to a More “Personalized” Approach to Cancer Staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- De Jong, M.C.; Nathan, H.; Sotiropoulos, G.C.; Paul, A.; Alexandrescu, S.; Marques, H.; Pulitano, C.; Barroso, E.; Clary, B.M.; Aldrighetti, L.; et al. Intrahepatic Cholangiocarcinoma: An International Multi-Institutional Analysis of Prognostic Factors and Lymph Node Assessment. J. Clin. Oncol. 2011, 29, 3140–3145. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.M.; Ribero, D.; O’Reilly, E.M.; Kokudo, N.; Miyazaki, M.; Pawlik, T.M. Intrahepatic Cholangiocarcinoma: Expert Consensus Statement. HPB 2015, 17, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Farges, O.; Fuks, D.; Boleslawski, E.; Le Treut, Y.-P.; Castaing, D.; Laurent, A.; Ducerf, C.; Rivoire, M.; Bachellier, P.; Chiche, L.; et al. Influence of Surgical Margins on Outcome in Patients with Intrahepatic Cholangiocarcinoma: A Multicenter Study by the AFC-IHCC-2009 Study Group. Ann. Surg. 2011, 254, 824–829. [Google Scholar] [CrossRef]
- Komuta, M.; Govaere, O.; Vandecaveye, V.; Akiba, J.; van Steenbergen, W.; Verslype, C.; Laleman, W.; Pirenne, J.; Aerts, R.; Yano, H.; et al. Histological Diversity in Cholangiocellular Carcinoma Reflects the Different Cholangiocyte Phenotypes. Hepatology 2012, 55, 1876–1888. [Google Scholar] [CrossRef]
- Neuzillet, C.; Casadei Gardini, A.; Brieau, B.; Vivaldi, C.; Smolenschi, C.; Brandi, G.; Tougeron, D.; Filippi, R.; Vienot, A.; Silvestris, N.; et al. Prediction of Survival with Second-Line Therapy in Biliary Tract Cancer: Actualisation of the AGEO CT2BIL Cohort and European Multicentre Validations. Eur. J. Cancer 2019, 111, 94–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Murakami, Y.; Uemura, K.; Sudo, T.; Hashimoto, Y.; Sasaki, H.; Sueda, T. Elevated Perioperative Serum CA 19-9 Levels are Independent Predictors of Poor Survival in Patients with Resectable Cholangiocarcinoma. J. Surg. Oncol. 2014, 110, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-K.; Lin, J.-J.; He, G.-H.; Wang, H.; Lu, J.-H.; Yang, G.-S. Preoperative Serum CA19-9 Levels is an Independent Prognostic Factor in Patients with Resected Hilar Cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 7890–7898. [Google Scholar] [PubMed]
- Loosen, S.H.; Roderburg, C.; Kauertz, K.L.; Koch, A.; Vucur, M.; Schneider, A.T.; Binnebösel, M.; Ulmer, T.F.; Lurje, G.; Schoening, W.; et al. CEA but Not CA19-9 is an Independent Prognostic Factor in Patients Undergoing Resection of Cholangiocarcinoma. Sci. Rep. 2017, 7, 16975. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Kummer, O.; Olschewski, M.; Otto, F.; Blum, H.E.; Opitz, O. Prognostic Relevance of Carbohydrate Antigen 19-9 Levels in Patients with Advanced Biliary Tract Cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2097–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gul, K.; Nas, S.; Ozdemir, D.; Gumus, M.; Ersoy, R.; Cakir, B. CA 19-9 Level in Patients with Type 2 Diabetes Mellitus and Its Relation to the Metabolic Control and Microvascular Complications. Am. J. Med. Sci. 2011, 341, 28–32. [Google Scholar] [CrossRef]
- Giannini, E.; Borro, P.; Botta, F.; Chiarbonello, B.; Fasoli, A.; Malfatti, F.; Romagnoli, P.; Testa, E.; Risso, D.; Lantieri, P.B.; et al. Cholestasis is the Main Determinant of Abnormal CA 19-9 Levels in Patients with Liver Cirrhosis. Int. J. Biol. Mark. 2000, 15, 226–230. [Google Scholar] [CrossRef]
- Tsen, A.; Barbara, M.; Rosenkranz, L. Dilemma of Elevated CA 19-9 in Biliary Pathology. Pancreatology 2018, 18, 862–867. [Google Scholar] [CrossRef]
- Lee, B.S.; Lee, S.H.; Son, J.H.; Jang, D.K.; Chung, K.H.; Paik, W.H.; Ryu, J.K.; Kim, Y.-T. Prognostic Value of CA 19-9 Kinetics during Gemcitabine-Based Chemotherapy in Patients with Advanced Cholangiocarcinoma. J. Gastroenterol. Hepatol. 2016, 31, 493–500. [Google Scholar] [CrossRef]
- Uenishi, T.; Yamazaki, O.; Tanaka, H.; Takemura, S.; Yamamoto, T.; Tanaka, S.; Nishiguchi, S.; Kubo, S. Serum Cytokeratin 19 Fragment (CYFRA21-1) as a Prognostic Factor in Intrahepatic Cholangiocarcinoma. Ann. Surg. Oncol. 2008, 15, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary Cancer: Utility of next-Generation Sequencing for Clinical Management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic Spectra of Biliary Tract Cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic Perturbations Reveal Distinct Regulatory Networks in Intrahepatic Cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and Prognostic Significance of EGFR, VEGF, and HER2 Expression in Cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and Genetic Characterization of Cholangiocarcinoma Identifies Therapeutic Targets for Tyrosine Kinase Inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Gong, Y.; Huang, F.; Lin, Q.; Zeng, B.; Li, Z.; Chen, R. Expression Levels and Significance of Nuclear Factor-ΚB and Epidermal Growth Factor Receptor in Hepatolithiasis Associated with Intrahepatic Cholangiocarcinoma. Dig. Surg. 2013, 30, 309–316. [Google Scholar] [CrossRef]
- Yang, X.; Wang, W.; Wang, C.; Wang, L.; Yang, M.; Qi, M.; Su, H.; Sun, X.; Liu, Z.; Zhang, J.; et al. Characterization of EGFR Family Gene Aberrations in Cholangiocarcinoma. Oncol. Rep. 2014, 32, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome Sequencing Identifies Frequent Inactivating Mutations in BAP1, ARID1A and PBRM1 in Intrahepatic Cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Govindan, A.; Sheth, R.A.; Nardi, V.; Blaszkowsky, L.S.; Faris, J.E.; Clark, J.W.; Ryan, D.P.; Kwak, E.L.; Allen, J.N.; et al. Prognosis and Clinicopathologic Features of Patients with Advanced Stage Isocitrate Dehydrogenase (IDH) Mutant and IDH Wild-Type Intrahepatic Cholangiocarcinoma. Oncologist 2015, 20, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, K.S.; Kang, K.J. Molecular Heterogeneity in Intrahepatic Cholangiocarcinoma. World J. Hepatol. 2020, 12, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Al Ustwani, O.; Iancu, D.; Yacoub, R.; Iyer, R. Detection of Circulating Tumor Cells in Cancers of Biliary Origin. J. Gastrointest. Oncol. 2012, 3, 97–104. [Google Scholar] [CrossRef]
- Yang, J.D.; Campion, M.B.; Liu, M.C.; Chaiteerakij, R.; Giama, N.H.; Ahmed Mohammed, H.; Zhang, X.; Hu, C.; Campion, V.L.; Jen, J.; et al. Circulating Tumor Cells are Associated with Poor Overall Survival in Patients with Cholangiocarcinoma. Hepatology 2016, 63, 148–158. [Google Scholar] [CrossRef]
- Correa-Gallego, C.; Maddalo, D.; Doussot, A.; Kemeny, N.; Kingham, T.P.; Allen, P.J.; D’Angelica, M.I.; DeMatteo, R.P.; Betel, D.; Klimstra, D.; et al. Circulating Plasma Levels of MicroRNA-21 and MicroRNA-221 are Potential Diagnostic Markers for Primary Intrahepatic Cholangiocarcinoma. PLoS ONE 2016, 11, e0163699. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Liu, X.; Zhang, Q.; Wang, C.; Zhao, Y. Diagnostic Value of MicroRNAs as Biomarkers for Cholangiocarcinoma. Dig. Liver Dis. 2016, 48, 1227–1232. [Google Scholar] [CrossRef]
- Fouassier, L.; Marzioni, M.; Afonso, M.B.; Dooley, S.; Gaston, K.; Giannelli, G.; Rodrigues, C.M.P.; Lozano, E.; Mancarella, S.; Segatto, O.; et al. Signalling Networks in Cholangiocarcinoma: Molecular Pathogenesis, Targeted Therapies and Drug Resistance. Liver Int. 2019, 39 (Suppl. 1), 43–62. [Google Scholar] [CrossRef] [Green Version]
- Cheon, Y.K.; Cho, Y.D.; Moon, J.H.; Jang, J.Y.; Kim, Y.S.; Kim, Y.S.; Lee, M.S.; Lee, J.S.; Shim, C.S. Diagnostic Utility of Interleukin-6 (IL-6) for Primary Bile Duct Cancer and Changes in Serum IL-6 Levels Following Photodynamic Therapy. Am. J. Gastroenterol. 2007, 102, 2164–2170. [Google Scholar] [CrossRef]
- Jørgensen, J.T.; Hersom, M. Companion Diagnostics-a Tool to Improve Pharmacotherapy. Ann. Transl Med. 2016, 4, 482. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region with Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Miyahara, K.; Nouso, K.; Tomoda, T.; Kobayashi, S.; Hagihara, H.; Kuwaki, K.; Toshimori, J.; Onishi, H.; Ikeda, F.; Miyake, Y.; et al. Predicting the Treatment Effect of Sorafenib Using Serum Angiogenesis Markers in Patients with Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Casadei Gardini, A.; Marisi, G.; Baron Toaldo, M.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma MiR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyöngyösi, B.; Végh, É.; Járay, B.; Székely, E.; Fassan, M.; Bodoky, G.; Schaff, Z.; Kiss, A. Pretreatment MicroRNA Level and Outcome in Sorafenib-Treated Hepatocellular Carcinoma. J. Histochem. Cytochem. 2014, 62, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Vaira, V.; Roncalli, M.; Carnaghi, C.; Faversani, A.; Maggioni, M.; Augello, C.; Rimassa, L.; Pressiani, T.; Spagnuolo, G.; Di Tommaso, L.; et al. MicroRNA-425-3p Predicts Response to Sorafenib Therapy in Patients with Hepatocellular Carcinoma. Liver Int. 2015, 35, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Negri, F.V.; Dal Bello, B.; Porta, C.; Campanini, N.; Rossi, S.; Tinelli, C.; Poggi, G.; Missale, G.; Fanello, S.; Salvagni, S.; et al. Expression of PERK and VEGFR-2 in Advanced Hepatocellular Carcinoma and Resistance to Sorafenib Treatment. Liver Int. 2015, 35, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Arao, T.; Ueshima, K.; Matsumoto, K.; Nagai, T.; Kimura, H.; Hagiwara, S.; Sakurai, T.; Haji, S.; Kanazawa, A.; Hidaka, H.; et al. FGF3/FGF4 Amplification and Multiple Lung Metastases in Responders to Sorafenib in Hepatocellular Carcinoma. Hepatology 2013, 57, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, E.; Stein, I.; Andreozzi, M.; Nemeth, J.; Shoham, A.; Pappo, O.; Schweitzer, N.; Tornillo, L.; Kanarek, N.; Quagliata, L.; et al. Human and Mouse VEGFA-Amplified Hepatocellular Carcinomas are Highly Sensitive to Sorafenib Treatment. Cancer Discov. 2014, 4, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Raoul, J.-L.; Adhoute, X.; Gilabert, M.; Edeline, J. How to Assess the Efficacy or Failure of Targeted Therapy: Deciding When to Stop Sorafenib in Hepatocellular Carcinoma. World J. Hepatol. 2016, 8, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Berhane, S.; Fox, R.; García-Fiñana, M.; Cucchetti, A.; Johnson, P. Using Prognostic and Predictive Clinical Features to Make Personalised Survival Prediction in Advanced Hepatocellular Carcinoma Patients Undergoing Sorafenib Treatment. Br. J. Cancer 2019, 121, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Caputo, F.; Dadduzio, V.; Tovoli, F.; Bertolini, G.; Cabibbo, G.; Cerma, K.; Vivaldi, C.; Faloppi, L.; Rizzato, M.D.; Piscaglia, F.; et al. The Role of PNI to Predict Survival in Advanced Hepatocellular Carcinoma Treated with Sorafenib. PLoS ONE 2020, 15, e0232449. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Cheng, A.-L.; Wyrwicz, L.; Ngan, R.; Blanc, J.-F.; Baron, A.D.; Vogel, A.; Ikeda, M.; Piscaglia, F.; et al. Analysis of Serum Biomarkers (BM) in Patients (Pts) from a Phase 3 Study of Lenvatinib (LEN) vs Sorafenib (SOR) as First-Line Treatment for Unresectable Hepatocellular Carcinoma (UHCC). Ann. Oncol. 2017, 28, v617. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Puzzoni, M.; Montagnani, F.; Marisi, G.; Tamburini, E.; Cucchetti, A.; Solaini, L.; Foschi, F.G.; Conti, F.; Ercolani, G.; et al. Profile of Lenvatinib in the Treatment of Hepatocellular Carcinoma: Design, Development, Potential Place in Therapy and Network Meta-Analysis of Hepatitis B and Hepatitis C in All Phase III Trials. Onco Targets Ther. 2019, 12, 2981–2988. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Park, J.O.; Ryoo, B.-Y.; Yen, C.-J.; Poon, R.; Pastorelli, D.; Blanc, J.-F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.F.; et al. Ramucirumab versus Placebo as Second-Line Treatment in Patients with Advanced Hepatocellular Carcinoma Following First-Line Therapy with Sorafenib (REACH): A Randomised, Double-Blind, Multicentre, Phase 3 Trial. Lancet Oncol 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated with Response to Regorafenib in Patients with Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [Green Version]
- Abuodeh, Y.; Naghavi, A.O.; Ahmed, K.A.; Venkat, P.S.; Kim, Y.; Kis, B.; Choi, J.; Biebel, B.; Sweeney, J.; Anaya, D.A.; et al. Prognostic Value of Pre-Treatment F-18-FDG PET-CT in Patients with Hepatocellular Carcinoma Undergoing Radioembolization. World J. Gastroenterol. 2016, 22, 10406–10414. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A Framework to Rank Genomic Alterations as Targets for Cancer Precision Medicine: The ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Brunetti, O.; Gnoni, A.; Licchetta, A.; Longo, V.; Calabrese, A.; Argentiero, A.; Delcuratolo, S.; Solimando, A.G.; Casadei-Gardini, A.; Silvestris, N. Predictive and Prognostic Factors in HCC Patients Treated with Sorafenib. Medicina 2019, 55, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + Ipilimumab (IPI) Combination Therapy in Patients (Pts) with Advanced Hepatocellular Carcinoma (AHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II Study of Durvalumab and Tremelimumab in Patients with Unresectable Hepatocellular Carcinoma (HCC): Phase I Safety and Efficacy Analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-Specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, Y.; Shi, C.; Zhou, X.; Xu, K.; Jiao, D.; Sun, Z.; Han, X. A Novel Immune Classification Reveals Distinct Immune Escape Mechanism and Genomic Alterations: Implications for Immunotherapy in Hepatocellular Carcinoma. J. Transl. Med. 2021, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of Established and Emerging Biomarkers of Immune Checkpoint Inhibitor Response in Advanced Hepatocellular Carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E. Role of ErbB Family Receptor Tyrosine Kinases in Intrahepatic Cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 7033–7058. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic Significance of Overexpression of C-Met Oncoprotein in Cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative Molecular Analysis of Intrahepatic Cholangiocarcinoma Reveals 2 Classes That Have Different Outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Cardinale, V.; Folseraas, T.; Overi, D.; Grzyb, K.; Costantini, D.; Berloco, P.B.; Di Matteo, S.; Karlsen, T.H.; Alvaro, D.; et al. Neoplastic Transformation of the Peribiliary Stem Cell Niche in Cholangiocarcinoma Arisen in Primary Sclerosing Cholangitis. Hepatology 2019, 69, 622–638. [Google Scholar] [CrossRef] [Green Version]
- Komuta, M.; Spee, B.; Vander Borght, S.; de Vos, R.; Verslype, C.; Aerts, R.; Yano, H.; Suzuki, T.; Matsuda, M.; Fujii, H.; et al. Clinicopathological Study on Cholangiolocellular Carcinoma Suggesting Hepatic Progenitor Cell Origin. Hepatology 2008, 47, 1544–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, Histomorphological and Molecular Classification of Cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 7–18. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast Growth Factor Receptor 2 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast Growth Factor Receptor 2 Translocations in Intrahepatic Cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate Dehydrogenase 1 and 2 Mutations in Cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boileve, A.; Baiev, I.; Dinicola, C.; Horick, N.K.; Tazdait, M.; Zhu, A.X.; Hollebecque, A.; Goyal, L. Clinical and Molecular Features of Patients with Cholangiocarcinoma Harboring FGFR Genetic Alterations. J. Clin. Oncol. 2019, 37, 4084. [Google Scholar] [CrossRef]
- Pignochino, Y.; Sarotto, I.; Peraldo-Neia, C.; Penachioni, J.Y.; Cavalloni, G.; Migliardi, G.; Casorzo, L.; Chiorino, G.; Risio, M.; Bardelli, A.; et al. Targeting EGFR/HER2 Pathways Enhances the Antiproliferative Effect of Gemcitabine in Biliary Tract and Gallbladder Carcinomas. BMC Cancer 2010, 10, 631. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.; Kasper, S.; Bitzer, M.; Block, A.; Sinn, M.; Schulze-Bergkamen, H.; Moehler, M.; Pfarr, N.; Endris, V.; Goeppert, B.; et al. PICCA Study: Panitumumab in Combination with Cisplatin/Gemcitabine Chemotherapy in KRAS Wild-Type Patients with Biliary Cancer-a Randomised Biomarker-Driven Clinical Phase II AIO Study. Eur. J. Cancer 2018, 92, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.H.; Chang, H.-M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Chang, J.S.; Jeung, H.C.; Kang, J.H.; et al. Gemcitabine and Oxaliplatin with or without Erlotinib in Advanced Biliary-Tract Cancer: A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardière, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.-F.; Viret, F.; et al. Gemcitabine and Oxaliplatin with or without Cetuximab in Advanced Biliary-Tract Cancer (BINGO): A Randomised, Open-Label, Non-Comparative Phase 2 Trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, C.; Chiang, N.J.; Tsai, C.S.; Tsou, H.H.; Huang, S.F.; Bai, L.Y.; Chang, I.C.; Shiah, H.S.; Ho, C.L.; et al. A KRAS Mutation Status-Stratified Randomized Phase II Trial of Gemcitabine and Oxaliplatin Alone or in Combination with Cetuximab in Advanced Biliary Tract Cancer. Ann. Oncol. 2015, 26, 943–949. [Google Scholar] [CrossRef]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular Pathways and Therapeutic Opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Silkin, S.V.; Startsev, S.S.; Krasnova, M.E.; Raskin, G.A.; Mitiushkina, N.V.; Iyevleva, A.G.; Sokolenko, A.P.; Imyanitov, E.N. Complete Clinical Response of BRAF-Mutated Cholangiocarcinoma to Vemurafenib, Panitumumab, and Irinotecan. J. Gastrointest. Cancer 2016, 47, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Lavingia, V.; Fakih, M. Impressive Response to Dual BRAF and MEK Inhibition in Patients with BRAF Mutant Intrahepatic Cholangiocarcinoma—2 Case Reports and a Brief Review. J. Gastrointest. Oncol. 2016, 7, E98–E102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loaiza-Bonilla, A.; Clayton, E.; Furth, E.; O’Hara, M.; Morrissette, J. Dramatic Response to Dabrafenib and Trametinib Combination in a BRAF V600E-Mutated Cholangiocarcinoma: Implementation of a Molecular Tumour Board and next-Generation Sequencing for Personalized Medicine. Ecancermedicalscience 2014, 8. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Lassen, U.N.; Elez, E.; Italiano, A.; Curigliano, G.; de Braud, F.G.; Prager, G.; Greil, R.; Stein, A.; Fasolo, A.; et al. Efficacy and Safety of Dabrafenib (D) and Trametinib (T) in Patients (Pts) with BRAF V600E–Mutated Biliary Tract Cancer (BTC): A Cohort of the ROAR Basket Trial. J. Clin. Oncol. 2019, 37, 187. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Boilève, A.; Verlingue, L.; Hollebecque, A.; Boige, V.; Ducreux, M.; Malka, D. Rare Cancer, Rare Alteration: The Case of NTRK Fusions in Biliary Tract Cancers. Expert. Opin. Investig. Drugs 2021, 1–9. [Google Scholar] [CrossRef]
- Patel, M.; Siena, S.; Demetri, G.; Doebele, R.; Chae, Y.; Conkling, P.; Garrido-Laguna, I.; Longo, F.; Rolfo, C.; Sigal, D.; et al. O-3 Efficacy and Safety of Entrectinib in NTRK Fusion-Positive Gastrointestinal Cancers: Updated Integrated Analysis of Three Clinical Trials (STARTRK-2, STARTRK-1 and ALKA-372-001). Ann. Oncol. 2020, 31, 232–233. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.; Salem, M.E.; Battaglin, F.; et al. Molecular Profile of BRCA-Mutated Biliary Tract Cancers. ESMO Open 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef]
- Tan, E.S.; Cao, B.; Kim, J.; Al-Toubah, T.E.; Mehta, R.; Centeno, B.A.; Kim, R.D. Phase 2 Study of Copanlisib in Combination with Gemcitabine and Cisplatin in Advanced Biliary Tract Cancers. Cancer 2020. [Google Scholar] [CrossRef]
- Silva, V.W.K.; Askan, G.; Daniel, T.D.; Lowery, M.; Klimstra, D.S.; Abou-Alfa, G.K.; Shia, J. Biliary Carcinomas: Pathology and the Role of DNA Mismatch Repair Deficiency. Chin. Clin. Oncol. 2016, 5, 62. [Google Scholar] [CrossRef]
- Diaz, L.A.; Le, D.; Maio, M.; Ascierto, P.A.; Geva, R.; Motola-Kuba, D.; André, T.; van Cutsem, E.; Gottfried, M.; Elez, E.; et al. Pembrolizumab in Microsatellite Instability High Cancers: Updated Analysis of the Phase II KEYNOTE-164 and KEYNOTE-158 Studies. Ann. Oncol. 2019, 30, v475. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Jain, A.; Shroff, R.T.; Zuo, M.; Weatherly, J.; Meric-Bernstam, F.; Isaacs, R.; Ali, S.M.; Bekaii-Saab, T.S.; Javle, M.M. Tumor Mutational Burden (TMB) and Co-Existing Actionable Mutations in Biliary Tract Cancers (BTC). J. Clin. Oncol. 2017, 35, 4086. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors with 10 or More Mutations per Megabase. JAMA Oncol. 2021. [Google Scholar] [CrossRef]
- Abdel-Wahab, R.; Ali, S.M.; Borad, M.J.; Shroff, R.T.; Kwong, L.; Vauthey, J.-N.; Koay, E.J.; Zuo, M.; Rashid, A.; Schrock, A.B.; et al. Variations in DNA Repair Genomic Alterations and Tumor Mutation Burden in Biliary Tract Cancer (BTC) Subtypes. J. Clin. Oncol. 2018, 36, 263. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.-Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and Safety of Pembrolizumab for the Treatment of Advanced Biliary Cancer: Results from the KEYNOTE-158 and KEYNOTE-028 Studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Renouf, D.; Lim, H. A Population Based Analysis of Prognostic Factors in Advanced Biliary Tract Cancer. J. Gastrointest. Oncol. 2014, 5, 428–432. [Google Scholar] [CrossRef]
- Tella, S.H.; Kommalapati, A.; Borad, M.J.; Mahipal, A. Second-Line Therapies in Advanced Biliary Tract Cancers. Lancet Oncol. 2020, 21, e29–e41. [Google Scholar] [CrossRef]
- Solimando, A.G.; Summa, S.D.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boilève, A.; Hilmi, M.; Delaye, M.; Tijeras-Raballand, A.; Neuzillet, C. Biomarkers in Hepatobiliary Cancers: What Is Useful in Clinical Practice? Cancers 2021, 13, 2708. https://doi.org/10.3390/cancers13112708
Boilève A, Hilmi M, Delaye M, Tijeras-Raballand A, Neuzillet C. Biomarkers in Hepatobiliary Cancers: What Is Useful in Clinical Practice? Cancers. 2021; 13(11):2708. https://doi.org/10.3390/cancers13112708
Chicago/Turabian StyleBoilève, Alice, Marc Hilmi, Matthieu Delaye, Annemilaï Tijeras-Raballand, and Cindy Neuzillet. 2021. "Biomarkers in Hepatobiliary Cancers: What Is Useful in Clinical Practice?" Cancers 13, no. 11: 2708. https://doi.org/10.3390/cancers13112708