
Whole Exome Sequencing of Biliary Tubulopapillary Neoplasms Reveals Common Mutations in Chromatin Remodeling Genes

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Clinical Information and Statistical Analyses
2.3. Tumor Samples
2.4. SNV Calling
2.5. CNV Analysis
2.6. Fluorescence In Situ Hybridization (FISH) Analysis of Chromosome 1
3. Results
3.1. Single Nucleotide Variants (SNVs)
3.2. Mutational Signature Pattern
3.3. Recurrently Mutated Genes and Corresponding Pathways
3.4. Identified Potential Therapeutic Targets
3.5. Genes Classically Associated with Pancreato-Biliary Carcinogenesis Are Rarely Altered in ITPN Carcinogenesis
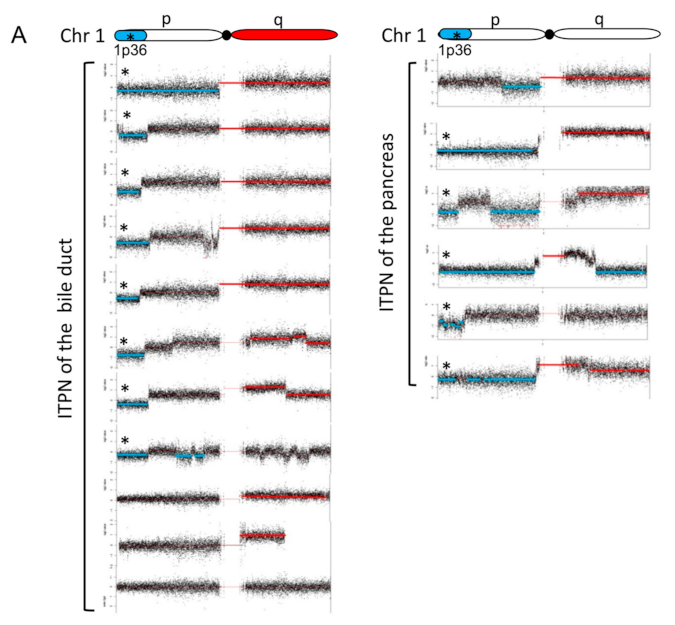
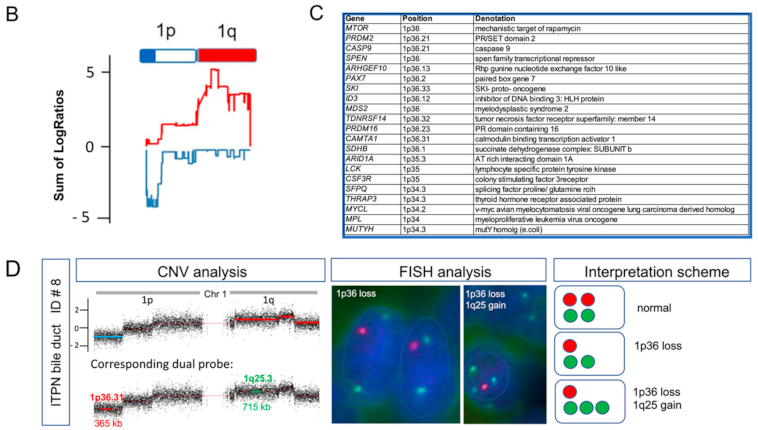
3.6. CNV Detects Recurrent Gains and Losses on Chromosome 1
3.7. Genetic Losses
3.8. Genetic Gains
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID # | Age at Diagnosis | Gender | Diagnosis | Invasive Tumor | Tumor Size (cm) | pT | pN | Survival (Months) | Status |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 75 | w | ITPN of the bile duct | no | 7.5 | n.a. | n.a | 31 | death |
| 2 | 59 | w | ITPN of the bile duct | yes | 6.0 | pT1 | pN0 (0/20) | 33 | alive at last follow-up |
| 3 | 48 | m | ITPN of the bile duct | yes (minimal invasive) | 15.0 | n.a. | n.a | 83 | alive at last follow-up |
| 4 | 72 | w | ITPN of the bile duct | yes | 6.7 | n.a. | pN0 (0/4) | 52 | alive at last follow-up |
| 5 | 68 | w | ITPN of the bile duct | yes | 5.5 | n.a. | pN0 (0/1) | 124 | alive at last follow-up |
| 6 | 64 | m | ITPN of the bile duct | yes | 9.0 | n.a. | n.a | 1 | perioperative death |
| 7 | 57 | w | ITPN of the bile duct | yes | 7.0 | pT2b | pN1 (1/24) | 39 | alive at last follow-up |
| 8 | 65 | w | ITPN of the bile duct | yes | 5.0 | n.a. | pN0 (0/4) | 55 | alive at last follow-up |
| 9 | 55 | w | ITPN of the bile duct | yes | 7.0 | n.a. | pN0 (0/5) | 1 | perioperative death |
| 10 | 58 | m | ITPN of the bile duct | yes | 5.0 | n.a. | n.a | 11 | alive at last follow-up |
| 11 | 78 | w | ITPN of the bile duct | yes | n.a. | n.a. | n.a | 150 | alive at last follow-up |
| 12 | 53 | m | ITPN of the pancreas | suspicious for invasion | 3.5 | pTis | pN0 (0/21) | 27 | alive at last follow-up |
| 13 | 87 | w | ITPN of the pancreas | no | 15.0 | pTis | pN0 | 33 | alive at last follow-up |
| 14 | 66 | w | ITPN of the pancreas | yes | 10.5 | pT2 | pN0 (0/24) | 3 | alive at last follow-up |
| 15 | 63 | m | ITPN of the pancreas | yes | 6.0 | pT3 | pN1 (1/17) | 70 | alive at last follow-up |
| 16 | 76 | m | ITPN of the pancreas | yes | 4.0 | pT3 | pN1 | 54 | alive at last follow-up |
| 17 | 63 | w | ITPN of the pancreas | yes | 1.8 | pT1c | pN1 (1/17) | 0 | perioperative death |
References
- Schlitter, A.M.; Jang, K.-T.; Klöppel, G.; Saka, B.; Hong, S.-M.; Choi, H.; Offerhaus, G.J.; Hruban, R.H.; Zen, Y.; Konukiewitz, B.; et al. Intraductal tubulopapillary neoplasms of the bile ducts: Clinicopathologic, immunohistochemical, and molecular analysis of 20 cases. Mod. Pathol. 2015, 28, 1249–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basturk, O.; Adsay, V.; Askan, G.; Dhall, D.; Zamboni, G.; Shimizu, M.; Cymes, K.; Carneiro, F.; Balci, S.; Sigel, C.; et al. Intraductal Tubulopapillary Neoplasm of the Pancreas: A Clinicopathologic and Immunohistochemical Analysis of 33 Cases. Am. J. Surg. Pathol. 2017, 41, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basturk, O.; Berger, M.F.; Yamaguchi, H.; Adsay, V.; Askan, G.; Bhanot, U.K.; Zehir, A.; Carneiro, F.; Hong, S.-M.; Zamboni, G.; et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Mod. Pathol. 2017, 30, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Katabi, N.; Torres, J.; Klimstra, D.S. Intraductal Tubular Neoplasms of the Bile Ducts. Am. J. Surg. Pathol. 2012, 36, 1647–1655. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Shimizu, M.; Ban, S.; Koyama, I.; Hatori, T.; Fujita, I.; Yamamoto, M.; Kawamura, S.; Kobayashi, M.; Ishida, K.; et al. Intraductal Tubulopapillary Neoplasms of the Pancreas Distinct From Pancreatic Intraepithelial Neoplasia and Intraductal Papillary Mucinous Neoplasms. Am. J. Surg. Pathol. 2009, 33, 1164–1172. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kuboki, Y.; Hatori, T.; Yamamoto, M.; Shiratori, K.; Kawamura, S.; Kobayashi, M.; Shimizu, M.; Ban, S.; Koyama, I.; et al. Somatic Mutations in PIK3CA and Activation of AKT in Intraductal Tubulopapillary Neoplasms of the Pancreas. Am. J. Surg. Pathol. 2011, 35, 1812–1817. [Google Scholar] [CrossRef]
- Hruban, R.H.; Adsay, N.V.; Esposito, I.; Fukushima, N.; Furukawa, T.; Klöppel, G.; Maitra, A.; Notohara, K.; Offerhaus, G.J.A.; Ohike, N.; et al. Pancreatic ductal adenocarcinoma. In WHO Classification of Tumours. Digestive System Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2019; pp. 322–332. [Google Scholar]
- Goeppert, B.; Toth, R.; Singer, S.; Albrecht, T.; Lipka, D.B.; Lutsik, P.; Brocks, D.; Baehr, M.; Muecke, O.; Assenov, Y.; et al. Integrative Analysis Defines Distinct Prognostic Subgroups of Intrahepatic Cholangiocarcinoma. Hepatology 2019, 69, 2091–2106. [Google Scholar] [CrossRef]
- Goeppert, B.; Renner, M.; Singer, S.; Albrecht, T.; Zhang, Q.; Mehrabi, A.; Pathil, A.; Springfeld, C.; Köhler, B.; Rupp, C.; et al. Prognostic Impact of Carboxylesterase 2 in Cholangiocarcinoma. Sci. Rep. 2019, 9, 4338. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, A.M.; Segler, A.; Steiger, K.; Michalski, C.W.; Jäger, C.; Konukiewitz, B.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci. Rep. 2017, 7, srep41064. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.; Engleitner, T.; Mueller, S.; Maresch, R.; Zwiebel, M.; González-Silva, L.; Schneider, G.; Banerjee, R.; Yang, F.; Vassiliou, G.S.; et al. Analysis pipelines for cancer genome sequencing in mice. Nat. Protoc. 2020, 15, 266–315. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.B.; Sougnez, C.; Gabriel, S.B.; Meyerson, M.L.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Kuilman, T.; Velds, A.; Kemper, K.; Ranzani, M.; Bombardelli, L.; Hoogstraat, M.; Nevedomskaya, E.; Xu, G.; De Ruiter, J.; Lolkema, M.P.; et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Pfarr, N.; Darb-Esfahani, S.; Leichsenring, J.; Taube, E.; Boxberg, M.; Braicu, I.; Jesinghaus, M.; Penzel, R.; Endris, V.; Noske, A.; et al. Mutational profiles of Brenner tumors show distinctive features uncoupling urothelial carcinomas and ovarian carcinoma with transitional cell histology. Genes Chromosom. Cancer 2017, 56, 758–766. [Google Scholar] [CrossRef]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.C.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.C.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signalling in cancer: Therapeutic targeting of Wnt signalling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Ladanyi, M.; Vega, F.S.; Zauderer, M. Loss of BAP1 as a candidate predictive biomarker for immunotherapy of mesothelioma. Genome Med. 2019, 11, 18. [Google Scholar] [CrossRef]
- Stumpf, E.; Aalto, Y.; Höög, A.; Kjellman, M.; Otonkoski, T.; Knuutila, S.; Andersson, L.C. Chromosomal alterations in human pancreatic endocrine tumors. Genes Chromosom. Cancer 2000, 29, 83–87. [Google Scholar] [CrossRef]
- El-Rifai, W.; Harper, J.C.; Cummings, O.W.; Hyytinen, E.R.; Frierson, H.F.; Knuutila, S.; Powell, S.M. Consistent genetic alterations in xenografts of proximal stomach and gastro-esophageal junction adenocarcinomas. Cancer Res. 1998, 58, 34–37. [Google Scholar]
- Savelieva, E.; Belair, C.D.; Newton, M.A.; Devries, S.; Gray, J.W.; Waldman, F.; A Reznikoff, C. 20q gain associates with immortalization: 20q13.2 amplification correlates with genome instability in human papillomavirus 16 E7 transformed human uroepithelial cells. Oncogene 1997, 14, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.-X.; Gu, Z.-H.; Han, B.; Zhang, R.-X.; Pan, C.-M.; Xiang, Y.; Rong, X.-J.; Chen, X.; Li, Q.-Y.; Wan, H.-Y. Whole-exome sequencing to identify novel somatic mutations in squamous cell lung cancers. Int. J. Oncol. 2013, 43, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Borad, M.J.; Gores, G.J.; Roberts, L.R. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Heining, C.; Horak, P.; Uhrig, S.; Codo, P.L.; Klink, B.; Hutter, B.; Fröhlich, M.; Bonekamp, D.; Richter, D.; Steiger, K.; et al. NRG1 Fusions in KRAS Wild-Type Pancreatic Cancer. Cancer Discov. 2018, 8, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, M.; Trujillo, M.A.; Huang, B.; Nanda, N.; Jiang, Z.; Jeong, Y.J.; Pflüger, M.; Goggins, M.G.; Hruban, R.H.; Thompson, E.D.; et al. Generation and characterization of a cell line from an intraductal tubulopapillary neoplasm of the pancreas. Lab. Investig. 2020, 100, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, C.; Carpentier, W.; Guettier, C.; Camilleri-Broët, S.; Borelli, W.V.; Dos Santos, C.R.C.; Castaing, D.; Duclos-Vallee, J.-C.; Broët, P. Patterns of chromosomal copy-number alterations in intrahepatic cholangiocarcinoma. BMC Cancer 2015, 15, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, A.; Mills, A.A. The Quest for the 1p36 Tumor Suppressor. Cancer Res. 2008, 68, 2551–2556. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.; Papazoglu, C.; Wu, Y.; Capurso, D.; Brodt, M.; Francis, D.; Bredel, M.; Vogel, H.; Mills, A.A. CHD5 Is a Tumor Suppressor at Human 1p36. Cell 2007, 128, 459–475. [Google Scholar] [CrossRef] [Green Version]
- Puri, L.; Saba, J. Getting a Clue from 1q: Gain of Chromosome 1q in Cancer. J. Cancer Biol. Res. 2014, 2, 1053. [Google Scholar]
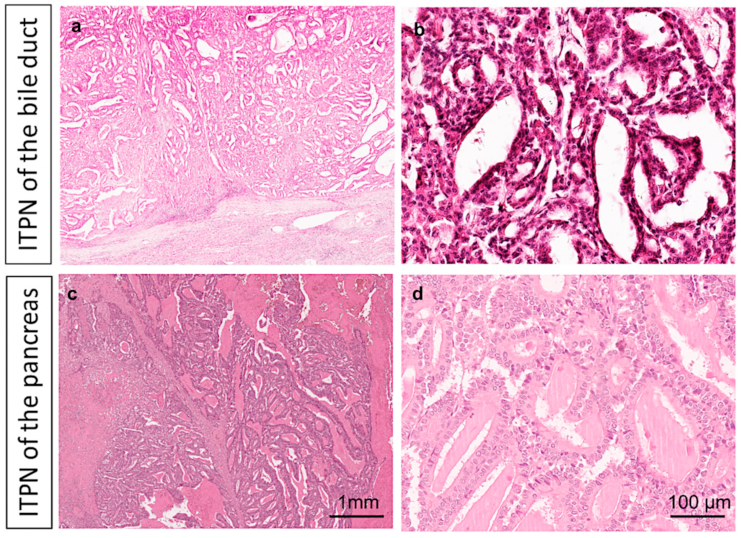
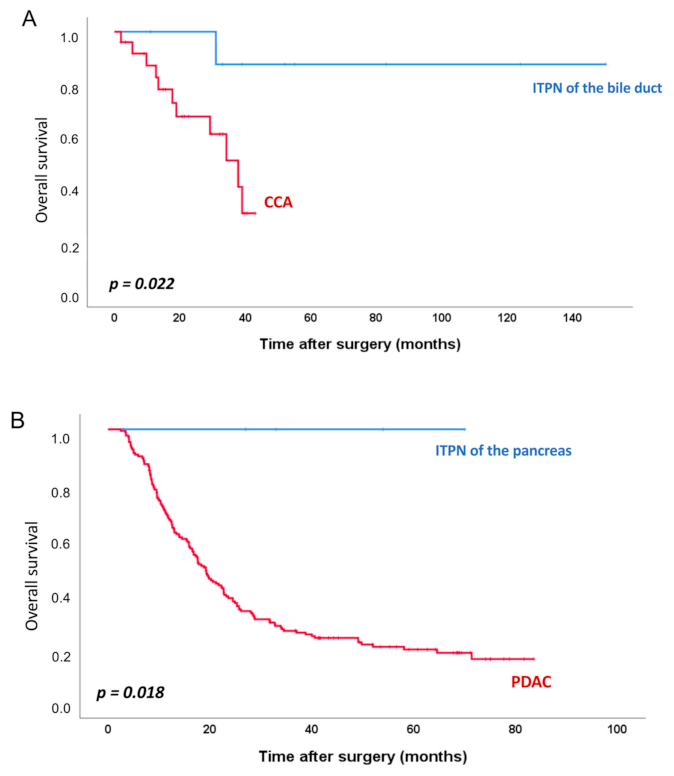
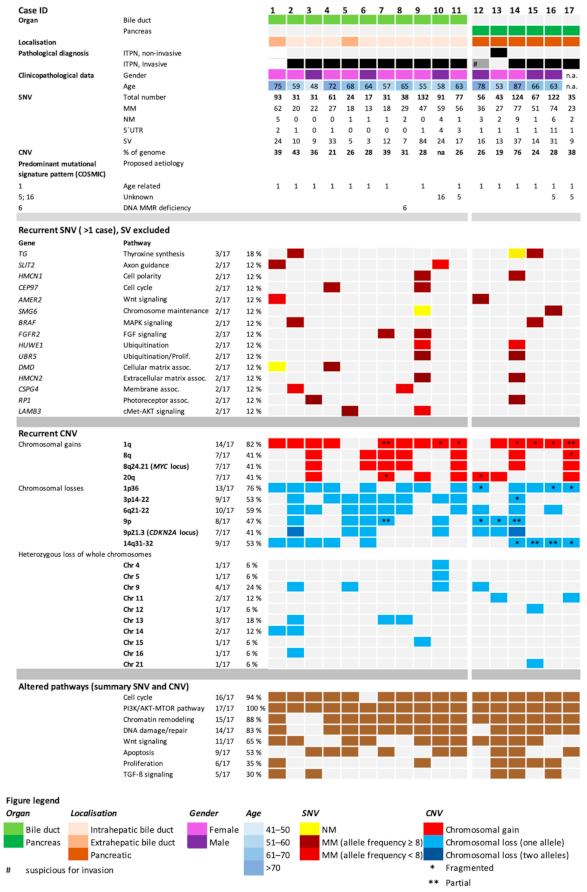
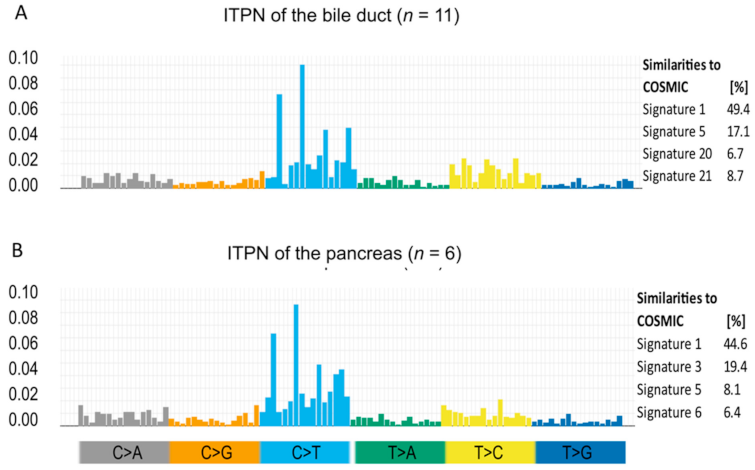

| Bile Duct | Pancreas | ||||
|---|---|---|---|---|---|
| Gene | Reported Mutation Rates in CCA [18] | Observed Frequency (SNV) in ITPN of the Bile Duct | Gene | Reported mutation Rates in PDAC [19] | Observed Frequency (SNV) in ITPN of the Pancreas |
| TP53 | 26% | 9% | KRAS | 92% | 0% |
| KRAS | 17% | 0% | TP53 | 45% | 0% |
| SMAD4 | 8% | 0% | SMAD4 | 17% | 17% |
| NF1 | 6% | 0% | FLG | 10% | 0% |
| ARID1A | 6% | 9% | ATXN1 | 7% | 0% |
| PBRM1 | 6% | 0% | COL14A1 | 6% | 0% |
| KMT2D | 6% | 0% | ITGAE | 6% | 0% |
| ATR | 6% | 0% | GLI3 | 6% | 0% |
| PIK3CA | 5% | 0% | GNAS | 6% | 0% |
| ERBB3 | 5% | 0% | SPTA1 | 6% | 0% |
| KMT2C | 5% | 9% | ARID1A | 5% | 0% |
| PIK3C2G | 4% | 0% | CDKN2A | 5% | 0% * |
| APC | 4% | 0% | RP1L1 | 5% | 0% |
| BAP1 | 4% | 9% | BCLAF1 | 5% | 0% |
| POLQ | 4% | 0% | AXIN1 | 5% | 0% |
| ARID2 | 4% | 9% | NIN | 5% | 0% |
| IDH1 | 3% | 0% | RNF43 | 4% | 0% |
| TET1 | 3% | 0% | RBM10 | 4% | 0% |
| CTNNB1 | 3% | 0% | IRF2 | 4% | 0% |
| BRAF | 3% | 9% | HDAC2 | 3% | 0% |
| Identified Actionable Genes | Case ID | OncoKB Search | |||||
|---|---|---|---|---|---|---|---|
| Pathway | Gene | Alteration | Entity | Drugs | Level of Evidence | Level-assoc. Cancer Types | |
| MAPK signaling | BRAF | p.Val600Glu | ITPN pancreas (1/6) | # 2 | Vemurafenib, Dabrafenib, Trametinib | 1 | Melanoma |
| Encorafenib + Cetuximab | 1 | Colorectal cancer | |||||
| NRAS | p.Gly12Asp | ITPN pancreas (1/6) | # 12 | Binimetinib, Combimetinib, Iodine I 131-6-Beta-Iodomethyl-19-Norcholesterol + Selumetinib | 3A | Melanoma, Histiocytosis, Thyroid cancer | |
| Panitumumab, Cetuximab | R1 | Colorectal cancer | |||||
| MTOR signaling | MTOR | p.Ser1863Leu* | ITPN pancreas (1/6) | # 14 | Everolimus, Temsirolimus | 4 | All solid tumors |
| FGF signaling | FGFR2 | p.Ile355Arg* p.Ile643Arg* | ITPN bile duct (2/11) | # 7,# 9 | Erdafitinib, Debio 1347, AZD4547, BGJ398 | 4 | All solid tumors |
| Chromatin remodeling | KDM6A | p.Ser440Leu* | ITPN bile duct (1/11) | # 8 | Tazemetostat | 4 | Bladder cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, C.; Engleitner, T.; Lange, S.; Weber, J.; Jesinghaus, M.; Konukiewitz, B.; Muckenhuber, A.; Steiger, K.; Pfarr, N.; Goeppert, B.; et al. Whole Exome Sequencing of Biliary Tubulopapillary Neoplasms Reveals Common Mutations in Chromatin Remodeling Genes. Cancers 2021, 13, 2742. https://doi.org/10.3390/cancers13112742
Gross C, Engleitner T, Lange S, Weber J, Jesinghaus M, Konukiewitz B, Muckenhuber A, Steiger K, Pfarr N, Goeppert B, et al. Whole Exome Sequencing of Biliary Tubulopapillary Neoplasms Reveals Common Mutations in Chromatin Remodeling Genes. Cancers. 2021; 13(11):2742. https://doi.org/10.3390/cancers13112742
Chicago/Turabian StyleGross, Claudia, Thomas Engleitner, Sebastian Lange, Julia Weber, Moritz Jesinghaus, Björn Konukiewitz, Alexander Muckenhuber, Katja Steiger, Nicole Pfarr, Benjamin Goeppert, and et al. 2021. "Whole Exome Sequencing of Biliary Tubulopapillary Neoplasms Reveals Common Mutations in Chromatin Remodeling Genes" Cancers 13, no. 11: 2742. https://doi.org/10.3390/cancers13112742