1. Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancer-associated diseases worldwide [
1], and it accounts for more than 80% of primary liver cancers [
2]. HCC poses a heavy disease burden, and the total number of liver cancer cases has been increasing with aging [
2]. HCC is associated with liver cirrhosis in approximately 80–90% of cases [
3]; patients with chronic viral hepatitis B and C infection have a high risk of HCC [
4]. The risk of HCC increases after the development of cirrhosis, increasing the likelihood of death due to liver failure or HCC [
5]. More than half a million individuals worldwide are annually diagnosed with HCC [
6]. Currently, there are limited treatment options for HCC. For the past two decades, the median survival time for patients with advanced HCC is less than one year, and the 5-year relative survival rate is below 9% [
7]. Patients with well-preserved and good liver function typically opt for surgical resection of the liver. However, the most effective treatment method to improve the survival of patients with HCC is liver transplantation [
8]. Unfortunately, this treatment often results in a poor prognosis, including a high risk of postoperative complications and tumor recurrence. Several strategies are available to extend the survival time of patients with liver cancer, such as transplantation; surgical resection; target drugs, such as sorafenib, lenvatinib, and regorafenib; and immunotherapy (nivolumab) [
9].
Janus kinases (Jaks) are nonreceptor tyrosine kinases encoded by the Jak gene. The Jaks family comprises four members, namely Jak1, Jak2, Jak3, and Tyk2. The Jak2 gene is a major component of the Jak-Stat signaling pathway, which is linked by interferon-responsive genes to signal transduction [
10,
11]. Jak2 was also found to participate in several different types of cancers and myeloproliferative diseases [
12,
13,
14,
15]. Currently, several important Jak2 inhibitors, such as ruxolitinib, pacritinib, fedratinib, and momelotinib, are available, and they differ from each other with respect to the inhibition of other kinases [
16]. Interestingly, in vitro treatment with a combination of chemotherapy and momelotinib is a potent inhibitor of Jak2; it suppressed CSCs-like cells and reduced tumor burden in a mouse model of human ovarian cancer [
17]. Epidemiological studies report that the hepatitis virus B and C (HBV and HCV) often were considered as the most common etiology of HCC, and these two viruses together were often known as one risk factor for HCC [
18]. Viral infection-associated HCC (vHCC) often develops resistance to sorafenib (a multi-kinase inhibitor) therapy [
19]. Thus, an important target that targets vHCC and overcomes drug resistance is required. Poly(ADP-ribose)polymerase (PARP)1 is an important member of the PARP family [
20], which upon activation promotes DNA repair in eukaryotes1. However, much evidence suggests PARP1 can go beyond DNA repair and may regulate many other key biological processes, such as chromatin strengthening, stress signaling, cell survival/death, inflammation, drug resistance, and differentiation. PARP1 also functions as a sensor of oxidative stress, and PARP1 inhibitors have been seen enhancing the cancer cell’s sensitivity towards chemotherapy. Many cell-based studies denoted that PARP1 interacts directly with STAT3 and mediated in vitro poly(ADP-ribosyl)action of STAT3 [
21]. However, the role of PARP1 linked with JAK-STAT signaling in drug resistant liver cancer has not been explored well.
In this study, we hypothesized that momelotinib could regulate the progression of HCC by targeting Jak2 and associated markers. Our study suggests that Jak2 and PAPR1 are expressed higher in vHCC than in non-viral infection-associated HCC (nvHCC) and the normal liver. Jak2 expression was correlated with poor prognosis and poor overall survival of patients with HCC. Treatment of momelotinib significantly inhibits Jak2, resulting in the reduction of the migratory, invasive property of vHCC cells. Interestingly, cell cycle arrest and inhibition of the stem cell-like phenotype of vHCC cells were also observed after the momelotinib treatment. Furthermore, the combined effect of momelotinib and sorafenib both at in vitro and in vivo synergistically suppresses the proliferation and targets the DNA repair enzyme of vHCC cells, and effectively reduces the tumor burden and therapy resistance.
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
We obtained 30 matched nvHCC, and vHCC samples as a kind gift from Dr. Wei-Hwa Lee, from the Department of Pathology, Taipei Medical University-Shuang Ho Hospital HCC tissue bank, following ethical approval for their use from the Institutional Review Board of the Taipei Medical University-Shuang Ho Hospital. The requirement for patients to sign informed consent was waived because tissue samples were obtained retrospectively from the Taipei Medical University-Shuang Ho Hospital HCC archive. This study was conducted in a cohort of patients with HCC cancer at Taipei Medical University Shuang-Ho Hospital, Taipei, Taiwan. The study was reviewed and approved by the institutional review board (TMU-JIRB: 201302016).
2.2. Cell Lines and Reagents
The human SNU-387 and SNU-475 HCC cell lines were purchased from American Tissue Culture Collection (ATCC). The cells were maintained under conditions recommended by the vendor, cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Thermo Fisher Scientific Inc., Waltham, MA, USA.) supplemented with 10% fetal bo-vine serum (FBS) and 1% penicillin-streptomycin (Invitrogen, Life Technologies, Saint-Louis, MO, USA) at 37 °C, in a 5% humidified CO
2 incubator. THLE-2 cell line was derived from primary normal liver cells by infection with SV40 large T antigen, HepG
2 was derived from a liver hepatocellular carcinoma both were purchased from ATCC. THLE-2 and HepG
2 cell lines were maintained in DMEM medium (Dulbecco’s Modified Eagle Medium; Gibco, Invitrogen), and all cells were incubated at 37 °C in a humidified incubator containing 5% CO
2 in air. Cells were sub-cultured at 80–90% confluency. The Jak inhibitor momelotinib (CYT387) [
21] and sorafenib (Catalog No. S7397), purchased from Selleck Chemicals, were dissolved in DMSO.
2.3. Microarray and RNAseq Pre-Processing and Analysis
Gene expression profiles, GSE14323, GSE14520, and GSE6764, were downloaded from the Gene Expression Omnibus (GEO) database (
www.ncbi.nlm.nih.gov/geo/, accessed on 4 May 2020), and the data of RNA-seq expression results containing samples of vHCC and nvHCC from patients with liver hepatocellular carcinoma (LIHC) were downloaded from TCGA portal Xena browser (
https://xenabrowser.net/, accessed on 10 May 2020) used for survival analysis. GEO dataset GSE62813 was used to identify sorafenib resistance in HCC.
2.4. Differential Expression Analysis
To determine differentially expressed genes between tumor and adjacent normal liver tissues in the GSE14520 cohort, the Bayes method and a linear model were employed using the R package limma. p values were adjusted using the false discovery rate controlling the Benjamini–Hochberg procedure. Genes with log2 fold change >1 and adjusted p values < 0.05 were considered significant.
2.5. Human Specimens and Immunohistochemistry (IHC)
Tumor tissues were harvested from patients with HCC. Samples were collected from the Department of Pathology, Shuang Ho Hospital, after obtaining written informed consent from patients (TMU-JIRB: 201302016). Clinical details of patients from the Taipei Medical University Shuang-Ho Hospital HCC cancer cohort shown in
Supplementary Table S1. Using virus infection as a distinguishing criterion,
Supplementary Table S1 contains 30 samples, which are divided into three groups, including normal tissues (10 samples), non-viral hepatocellular carcinoma (10 samples), and viral hepatocellular carcinoma (10 samples). The collected samples were fixed in 4% paraformaldehyde and embedded in paraffin, with 5-µm sections cut from paraffin blocks. Staining was performed using anti-Jak2 (1:500; cat. ab39636, Abcam), followed by staining using the secondary antibody and H&E. The percentage of stained area to the selected field was recorded in a 5% interval, ranging from 0% to 100%. The staining intensity was graded into 3 categories (absent or weak, 1; moderate, 2; strong, 3). Q-score was derived from the product of percentage (P) of tumor cells with characteristic IHC staining (0–100%) and the intensity (I) of IHC staining (1-3) (Q = P × I; maximum = 300).
2.6. Cell Viability Test and Calculation of the Combination Index
The stock of momelotinib and sorafenib was prepared by dissolving 20 mg/mL of the mixture in DMSO. The stocks of each drug were stored at −20 °C until use. Using the CompuSyn software to calculate the half-maximal inhibitory concentration (IC
50) values of different cell lines as previously reported by Chou TC and Martin N. The calculation method of IC
50 is as described in the PC Software and User’s Guide on the ComboSyn Inc. website (
http://www.combosyn.com, accessed on 4 May 2020). The effects of momelotinib and sorafenib on cell proliferation were detected using the sulforhodamine B (SRB) assay. The synergistic effect of these two drugs was analyzed using isobolograms of the drug combination, as previously reported by Chou and Talalay [
22,
23]. The interaction between the two drugs was also analyzed by the median-effect principle proposed by them. The combination index (CI) was calculated using CompuSyn software. A CI value is less than 1 represented synergism [
23]. Briefly, HCC cells or tumorspheres were seeded in 96-well plates (8 × 10
5 cells/well) and treated with the drugs (momelotinib or sorafenib alone or in combination) at the 5 μM and 2.5 μM concentration for 48 h, respectively. After respective drug treatments, the relative cell number was estimated by the SRB reagent according to the manufacturer’s protocol (Sigma, Vallejo, CA, USA).
2.7. Transient Transfection and Dual-Luciferase Assay
SNU-387 and SNU-475 cells were seeded at a density of 1 × 106 cells in 100-mm2 culture plates. On the following day, cells were treated with TransFectin (Bio-Rad Laboratories, CA, USA) and transfected with 2 µg of pJAK2-TA-Luc and 2 µg of pRL-TK (Renilla luciferase control reporter plasmid (Promega, Foster, CA, USA). After 5 h of transfection, cells were trypsinized and seeded onto sterile, black-bottomed 96-well plates at a density of 1 × 104 cells per well and then incubated with the complete medium for 24 h. Cells were treated with either test compounds or 0.1% DMSO for 24 h. After treatment, cells were harvested in 20 µL of passive lysis buffer, and luciferase activity was evaluated using the dual-luciferase reporter assay kit (Promega) on Wallac 1420 VICTOR2 (PerkinElmer, Inc., Los Angeles, CA, USA). The experiments were performed in triplicate and repeated thrice. Relative luciferase activity was calculated according to the following formula: relative luciferase activity (%) = ([normalized luciferase activity of sample treated with a test compound]/[normalized luciferase activity of sample treated with 0.1% DMSO]) × 100.
2.8. Apoptosis and Cell Cycle Analysis
Apoptosis and cell cycle were analyzed through flow cytometry (Beckman, Fullerton, CA, USA) using the Annexin V/7AAD (FITC-conjugated) apoptosis kit (F-6012; US Everbright Inc., San Ramon, CA, USA) or propidium iodide (PI) according to the manufacturer’s protocol. To determine the effect on the cell cycle, HCC cells were exposed to momelotinib for 48 h. Thereafter, cells were washed and fixed with 70% ethanol. The cells were washed, re-suspended, and stained with 10 µg/mL of PI in PBS for 30 min at room temperature in the dark. The cells were analyzed through flow cytometry (Becton Dickinson, Mountain View, CA, USA), and the population of cells in each phase was counted.
2.9. TUNEL Assay
Cell death (apoptosis) in the tumor tissue was detected by staining HCC tissues using an in situ cell death detection kit from Roche according to the manufacturer’s protocol. Quantification was performed by calculating the percentage of TUNEL-positive cells by using a fluorescence microscope. The results are expressed as the mean number of TUNEL-positive apoptotic HCC cells in each group.
2.10. ALDEFLUOR Assay and ALDH1+ Cell Sorting by FACS
ALDH activity in HCC cells was assayed using the ALDEFLUOR kit according to the manufacturer’s instructions (STEMCELL Technologies, Durham, NC, USA). The cells that showed positive activity of aldehyde dehydrogenase 1 (ALDH1) were isolated and analyzed. Briefly, the human HCC cell lines SNU-387 was suspended at a concentration of 1 × 106 cells/mL in ALDEFLUOR assay buffer containing the ALDH substrate (BAAA, 1 μmol/L per 1 × 106 cells) and incubated for 40 min at 37 °C. Cells incubated with ALDEFLUOR substrate and treated with 50 mmol/L of diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor, were used as a reference control. To avoid contamination of cells of mouse origin from xenotransplanted tumors, they were stained with the anti-H2Kd antibody (BD Biosciences, 1/200, 20 min on ice) and then with a secondary antibody labeled with phycoerythrin (Jackson Labs, 1/250, 20 min on ice). Cells stained with PI alone were used as negative controls, and ALDEFLUOR-stained cells treated with DEAB and those stained with the secondary antibody alone were considered viable.
2.11. Western Blotting and qRT-PCR
Cells were washed with PBS and then lysed in RIPA lysis buffer; cellular protein lysates were isolated using the protein extraction kit (QIAGEN, Portland, OR, USA) and quantified using the Bradford protein assay kit (Bio-Rad, Taiwan). Approximately 20 μg of the sample from different experiments were loaded and subjected to SDS-PAGE by using the Mini-PROTEAN III system (Bio-Rad, Taipei, Taiwan). Separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane by using the Trans-Blot Turbo Transfer System (Bio-Rad), followed by blocking with Tris-buffered saline plus skimmed milk. These PVDF membranes were then probed with respective primary antibodies, followed by the secondary antibody. The commercial antibodies are shown in
Supplementary Table S2. The enhanced chemiluminescence detection kit was used to detect the proteins of interest. Images were captured and analyzed using the UVP BioDoc-It system (Upland, CA, USA). qRT-PCR was performed by using isolating total RNA using TRIzol-based protocol (Life Technologies, San Jose, CA, USA) provided by the manufacturer. In brief, one µg of total RNA was reverse transcribed using QIAGEN OneStep RT-PCR kit (QIAGEN, Taipei, Taiwan), and PCR was performed using a Rotor-Gene SYBR Green PCR kit (QIAGEN, Tiapei, Taiwan). The primer sequences are shown in
Supplementary Table S3.
2.12. Colony Formation Assay
The colony formation assay was performed according to a previously explained protocol [
24] with modifications. Briefly, a total of 500 colon cancer cells were seeded in six-well plates and treated with momelotinib. The cells were allowed to grow for another week and then harvested, fixed, and counted.
2.13. Wound Healing Migration Assay
Cells were seeded in six-well plates (Corning, New York, NY, USA) with RPMI 1640 medium containing 10% FBS and cultured to 95–100% confluence. A scratch along the median axis was then made with a sterile yellow pipette tip across cells. Cell migration pictures were captured at 0 and 48 h after the medium scratch, under a microscope, and analyzed using the NIH Image J software. (
https://imagej.nih.gov/ij/download.html, accessed on 4 May 2020).
2.14. Matrigel Invasion Assay
Cells (2 × 105) were seeded in 24-transwell chambers with an 8-μm pore membrane coated with Matrigel in the upper chamber of the transwell system containing serum-free RPMI 1640 medium. The lower chamber of the transwell system contained the medium with 20% FBS. After incubation at 37 °C for 6 h, non-invaded HCC cells on the upper side of the membrane were carefully removed with a cotton swab, and the invaded cells were stained with crystal violet dye, air-dried, and photographed under a microscope. Images were analyzed using the NIH Image J software.
2.15. Sphere Formation Assay
Cells (5 × 103/well) were plated in ultra-low-attachment six-well plates (Corning) containing stem cell medium consisting of serum-free RPMI 1640 medium supplemented with 10 ng/mL of human basic fibroblast growth factor (bFGF; Invitrogen, Grand Island, NY, USA), 1 × B27 supplement, and 20 ng/mL epidermal growth factor (EGF; Invitrogen). The medium was changed every 72 h. After 14 days of incubation, the formed spheres were counted and photographed.
2.16. Animal Studies
All animal experiments and maintenance were in strict compliance with the Animal Use Protocol Taipei Medical University (protocol LAC-2019-0526). The anti-proliferative effect of momelotinib and sorafenib in combination with vHCC cells in vivo investigated, athymic nude mouse models bearing HCC cell xenografts were established. Five-week-old male athymic nude mice were used for this study. The animal experiment is set to six mice per group. The mice were maintained under pathogen-free conditions and were provided with sterilized food and water. First, 1 × 106 SNU-387 cells were subcutaneously injected into the right flank near the hind leg of each nude mouse. When the mice had palpable tumors (tumor volume of approximately 100 mm3), they were randomly divided into control (100 µL of normal saline [NS] by intraperitoneal injection plus 100 µL of 1% DMSO) and 0.5% carboxymethyl cellulose ([CMC]-Na sterile water), momelotinib (30 mg/kg/day by intraperitoneal injection plus 100 µL of 1% DMSO and 0.5% CMC-Na sterile water), sorafenib (30 mg/kg/day by intragastric administration plus 100 µL NS by intraperitoneal injection), and combination (momelotinib, 30 mg/kg/day by intraperitoneal injection plus sorafenib 30 mg/kg/day by intragastric administration) groups (n = 6 animals/group). The treatments were performed 5 times/week for 4 weeks. The tumor volume was detected every week and was calculated using the following formula: volume (V) = π/6 × length × width × height. After 4 weeks, mice were humanely euthanized, and the tumors were isolated for further analyses.
2.17. Statistical Analysis
All assays were performed at least thrice in triplicate. Values are expressed as the mean ± standard deviation (SD). Comparisons between groups were estimated using Student’s t-test for cell line experiments or the Mann–Whitney U-test for clinical data, Spearman’s rank correlation between variables, and the Kruskal–Wallis test for comparison of three or more groups. The Kaplan–Meier method was used for the survival analysis, and the difference between survival curves was tested by a log-rank test. Univariate and multivariate analyses were based on the Cox proportional hazards regression model. All statistical analyses were performed using IBM SPSS Statistics for Windows, version 20 (IBM, Armonk, NY, USA). A p-value < 0.05 was considered statistically significant.
4. Discussion
The growing HCC is one of the leading causes of cancer-associated deaths worldwide. In more than 80–90% of cases, HCC is directly or indirectly associated with liver cirrhosis; chronic viral hepatitis B and C infections are mainly responsible for the development of HCC [
1,
2,
3,
4,
5]. Often this viral infection-associated with the development of resistance to sorafenib (a multi-kinase inhibitor) therapy [
20]. The overall 5 years survival rate of HCC is below 9% [
8]. Janus kinases (Jaks) are nonreceptor tyrosine kinases encoded by the Jak gene. The Jaks family comprises four members, namely Jak1, Jak2, Jak3, and Tyk2 [
22]. Jak2 has a role in several different types of cancers and myeloproliferative diseases [
13,
14,
15,
16]. Significantly increased expression of phosphorylated Jak2 and STAT3 are observed in Vhcc [
23]. Patients with this disease develop resistance to sorafenib, a multi-kinase inhibitor. Currently, several important Jak2 inhibitors are available, such as ruxolitinib, pacritinib, fedratinib, and momelotinib [
17], each of which has a different mode of action. Interestingly, the combination of chemotherapy and momelotinib is a potent inhibitor of Jak2, and it suppresses CSC-like cells and reduces tumor burden in vHCC [
18]. Thus, finding an important target that re-sensitizes and overcomes the drug resistance of vHCC is required.
The Janus kinase signal transducer and activator of transcription (JAK-STAT) signal is essential for a variety of cellular processes including survival, differentiation and proliferation. Currently, four therapeutic JAK2 inhibitors (ruxatinib, melatinib, momolotinib and pacomatinib) have been approved or are in advanced clinical studies. Molotinib (CYT387) is a dual inhibitor of JAK1 and JAK2 [
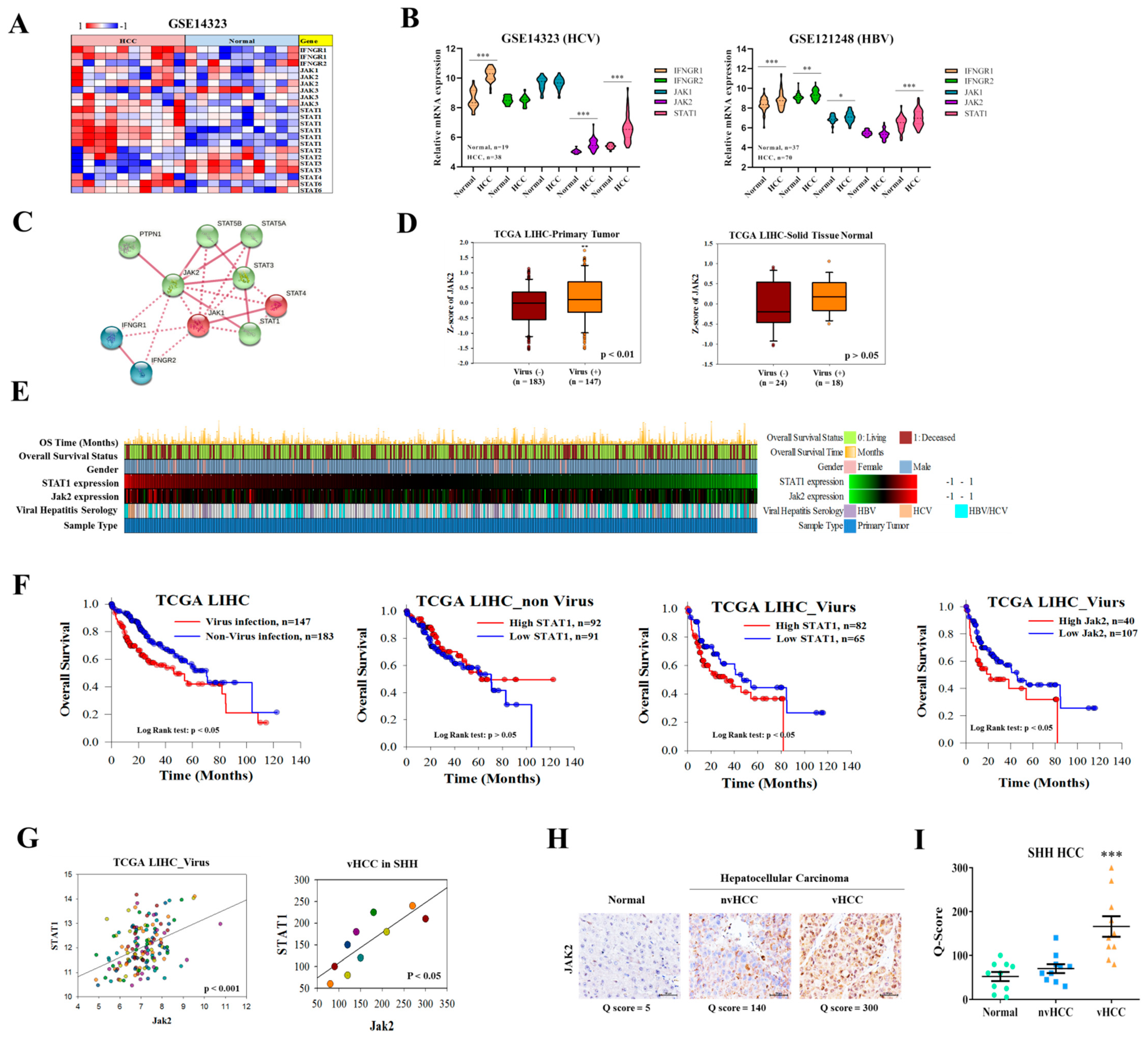
24]. The related downstream JAK1 and JAK2 in the IFNGR pathway induced by viral infection are related. Hence, we selected this dual inhibitor to evaluate its therapeutic effect. In addition, this is the first study to explore the application of Molotinib to viral liver cancer. In the present study, we first demonstrated that the expression of Jak1/2 and PAPR1 is significantly upregulated in vHCC than in nvHCC/normal liver tissues (
Figure 1 and
Figure 6). In addition, the proteins of IFN gamma-related pathways are activated after virus infection (
Figure 1B). This result suggests the difference in signal transmission between viral liver cancer and general liver cancer. Based on this observation, we are linked to the relevant targets of the JAK family and the potential applications of targeted therapy inhibitors. To further confirm our hypothesis that the JAK family and virus infection cause cancer. We also analyzed the overall survival between vHCC and HCC. Among them, STAT1 expression in both is a significant difference. In virus infection, the performance of IFN-gamma is an important indicator. IFN-gamma binds to nearby uninfected cell membrane receptors, stimulating signaling pathways to interfere with virus replication; stimulating cells to hydrolyze pathogenic proteins to prevent cells from being infected by the same or different viruses. In addition, STAT1 is a downstream gene of JAK2 in IFN-gamma signaling pathways, which can infer the significance of JAK2 in viral infection. Previous research indicated that overall Jak2 expression is related to the overall survival in HCC patients [
25]. Thus, the Jak family gene might be playing a key role in patients with HCC. Verstovsek et al. [
26] demonstrated the efficacy of momelotinib as a potent inhibitor of Jak1 and Jak2 in patients with primary and secondary myelofibrosis. The inhibitory effect of momelotinib (CYT387) on JAK2 phosphorylation is consistent with previous reports [
26]. Momelotinib (CYT387) is an ATP-competitive small molecule that potently inhibits JAK1/JAK2 kinases. Furthermore, we found that momelotinib significantly inhibited the growth of HCC cells (
Figure 2) and inhibit the phosphorylation of Jak2 and STAT3 and reduce the expression of p-Jak2, which verified the importance of momelotinib in targeting Jak2 and reducing tumorigenesis in HCC. Often HCC patients show sorafenib therapy resistance [
27]. Drug resistance is a major challenge in anticancer therapy. CSCs provide an alternative explanation for the aforementioned therapeutic challenges of several cancers [
28]. This small population of cancer cells has stem cell-like features such as tumorigenicity, self-renewal, and more resistance to chemotherapeutic agents than that shown by cancer cells [
29,
30]. Treatment of vHCC with momelotinib reduced the expression of cancer stemness markers, such as CD133, KLF4, and SOX2, and decreased ALDH1 activity (
Figure 4), suppressing tumorsphere and colony formation of HCC cells.
In the previous series of experiments, it has been preliminarily confirmed that the process of viral liver cancer may be related to the IFNGR-JAK-STAT pathway. Therefore, momelotinib was chosen as a JAK1/2 target combination therapy inhibitor to further prove our hypothesis. Momelotinib competes with JAK1/2 for ATP binding, which may result in inhibition of JAK1/2 activation, inhibition of the JAK-STAT signaling pathway, and so the induction of apoptosis and a reduction of tumor cell proliferation in JAK1/2-expressing tumor cells. Combination therapy, a treatment modality that combines two drugs for cancer therapy, has gained wide acceptance and popularity. It combines two drug targets involved in multiple pathways in cancer, utilizing different mechanisms to reduce the development of drug resistance in tumors [
31]. Combinations of two therapeutic agents into a biological system may produce a synergistic effect, or, sometimes, antagonistic, or identical effects, compared with their effects when acting separately. Momelotinib, together with sorafenib, increased the sensitivity of HCC cells, resulting in decreased colony formation in cells treated with the combination, and also alleviated apoptotic markers (Annexin-V+ and 7-AAD positive cells) PARP-1, PDCD4, and Bax molecules (
Figure 5). The integrity of the genome relies on cell cycle control and DNA repair to work together. PARP1 also functions as a sensor of oxidative stress, and PARP1 inhibitors have been seen enhancing the cancer cell’s sensitivity towards chemotherapy.
Since the role of PARP1 in DNA repair must be completed in the nucleus, it is extremely important to understand how to regulate the distribution of PARP1 in the cell. Furthermore, PARP1 inhibitors have become increasingly important in the field of cancer treatment. Considering the resistance of current PARP1 activity inhibitors, the use of regulating its cell distribution may also achieve the effect of inhibiting PARP1.
Figure 6 shows PARP1 expression associated with poor liver cancer patients’ overall survival. Based on the results of these analyses, we have concluded that a novel path in which momelotinib induces DNA damage and sensitizes viral hepatitis associated hepatocellular carcinoma aggressiveness and stemness via IFNGR-JAK2-STAT1-PARP1 axis. We hope that this discovery can develop possible PARP1 nuclear metastasis inhibitors and test its possibility as cancer treatment drugs. We hope that through this research, we can further analyze the correlation between cell cycle control and DNA repair, and provide another type of PARP1 activation mechanism for cancer treatment strategies.
Similarly, the combination of momelotinib and sorafenib efficiently suppressed the tumor-initiating ability in xenograft models (
Figure 7) and effectively inhibited the expression of Ki-67, a marker for representing tumor proliferation. Moreover, momelotinib facilitated sorafenib-induced apoptosis in xenograft tumors, as evaluated by the cleaved caspase-3 staining and TUNEL assay. Despite the fact that treatment with sorafenib is a useful therapeutic way for HCC, the survival rate of patients is still limited. This is because that HCC cells are heterogeneous cells with incongruous activation of several signaling pathways [
32,
33]. Combination therapy strategies have been proposed to improve the efficacy of sorefenib based treatment of HCC patients in many studies [
34,
35]. HCC patients treated with a combination therapy strategy resulted in more effective treatment, such as mTOR inhibitors and monoclonal antibodies. The JAK/STAT is an important pathway for cellular functions, including cell proliferation and differentiation, and it also participates in the mechanism of liver regeneration and gluconeogenesis [
36]. Many types of research indicated that the JAK/STAT pathway is often deregulated in signaling in HCC and other cancer [
37,
38]. The common JAK inhibitors including pacritinib, cryptotanishinone, and ruxolitinib have been used for cancer and human-relevant disease studies. Recently, Justin Jit Hin Tang et al. (2020) discussed the JAK/STAT signaling in hepatocellular carcinoma and organized different JAK/STAT inhibitors used in targeting the JAK/STAT pathway for HCC treatment, including small molecule inhibitors and siRNAs [
39]. In our report, the overexpression of Jak2 was suppressed by momelotinib, a Jak inhibitor, leading to a sharp reduction in the cell viability and DNA repair of vHCC cell lines. It has a wider significance, and this model may lead to a new therapeutic strategy for vHCC and non-vHCC.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}