
Clinicopathological-Associated Regulatory Network of Deregulated circRNAs in Hepatocellular Carcinoma

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. HCC Patients and Clinical Samples
2.2. Workflow for Analyses of Clinically Relevant circRNA Networks in HCC
3. Results
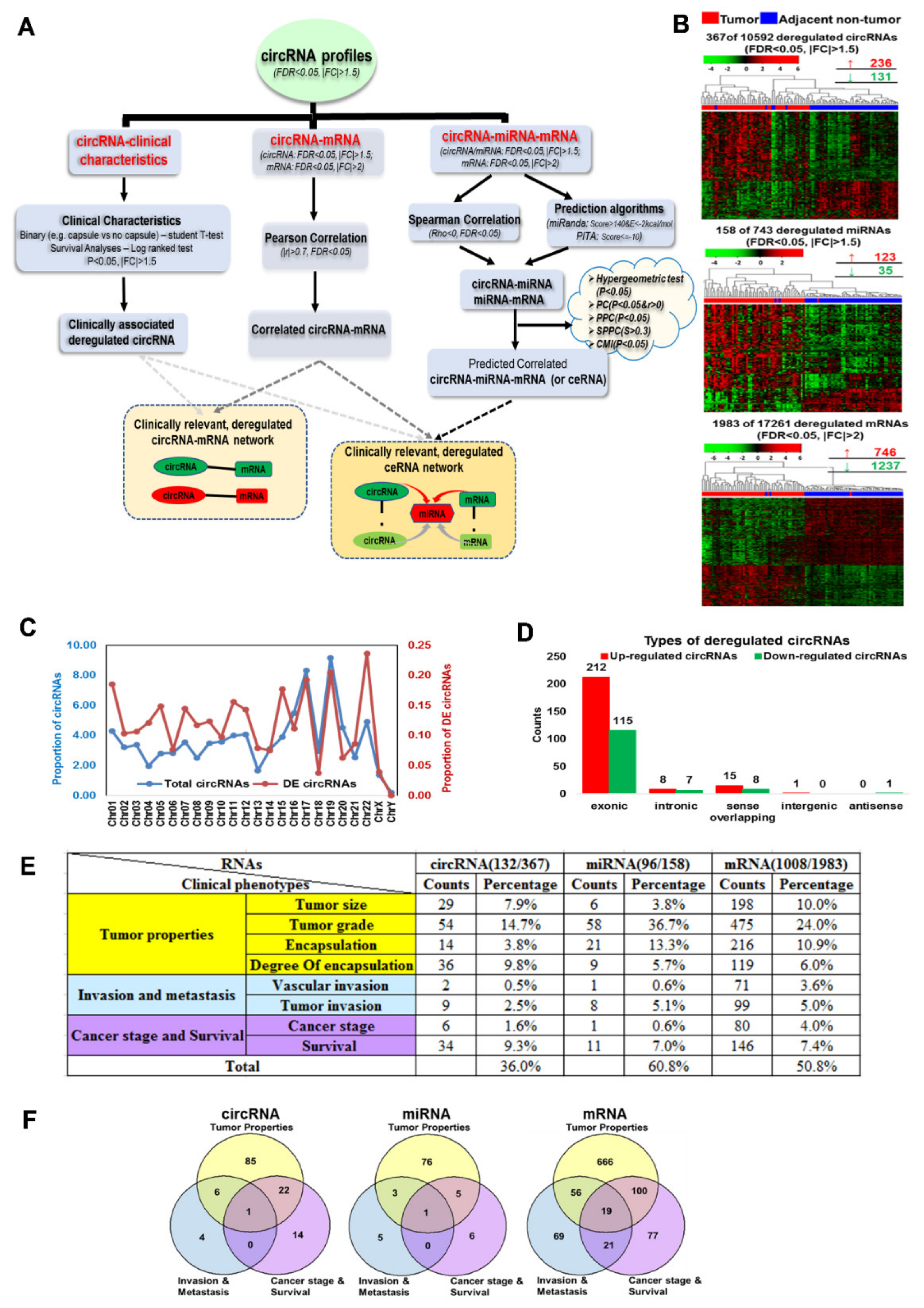
3.1. Profiles of Differentially Expressed circRNAs, miRNAs and mRNAs in HCC Patients
3.2. Deregulated circRNAs, miRNAs and mRNAs Are Associated with Various Clinicopathological Features
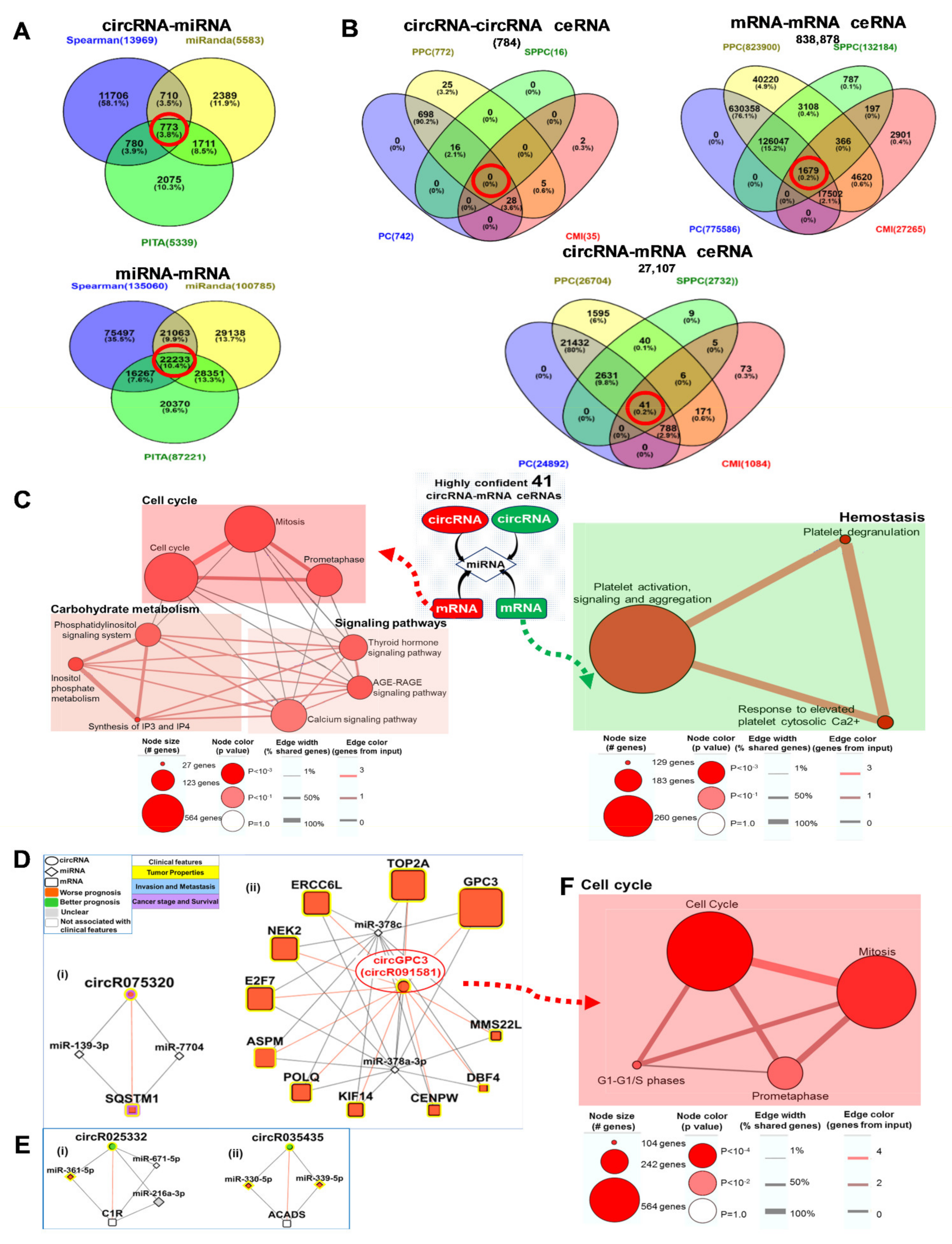
3.3. Clinically Relevant Deregulated Co-Expressed circRNA-mRNA Networks
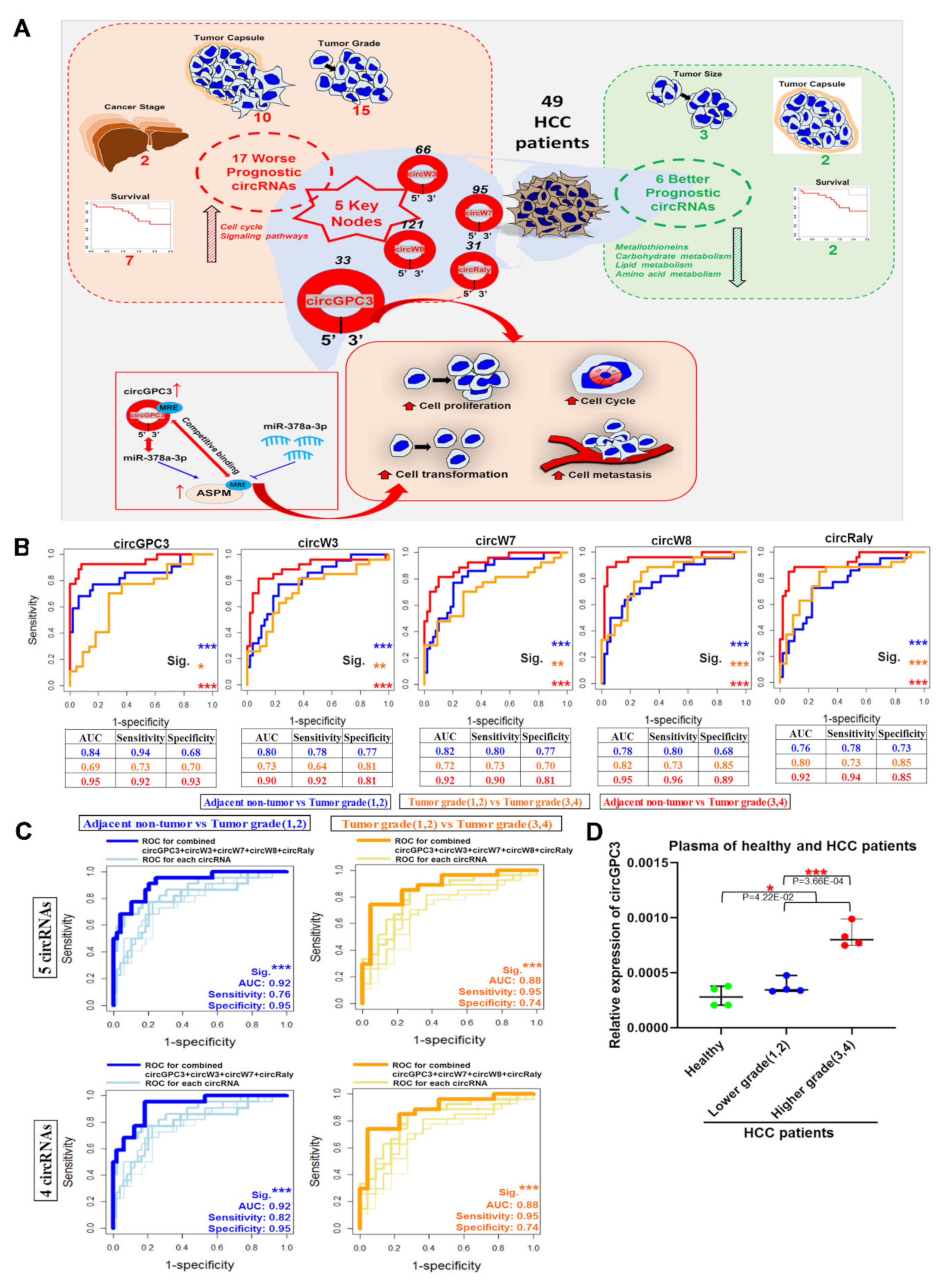
3.4. Oncogenic Roles of the Five Nodal/Master circRNAs Associated with Worse Clinical Characteristics
3.5. The Five Nodal circRNAs Modulate Tumorigenesis by Promoting Proliferation and Anchorage Independent Growth
3.6. Clinically Relevant Differentially Expressed circRNA-miRNA-mRNA ceRNA Networks
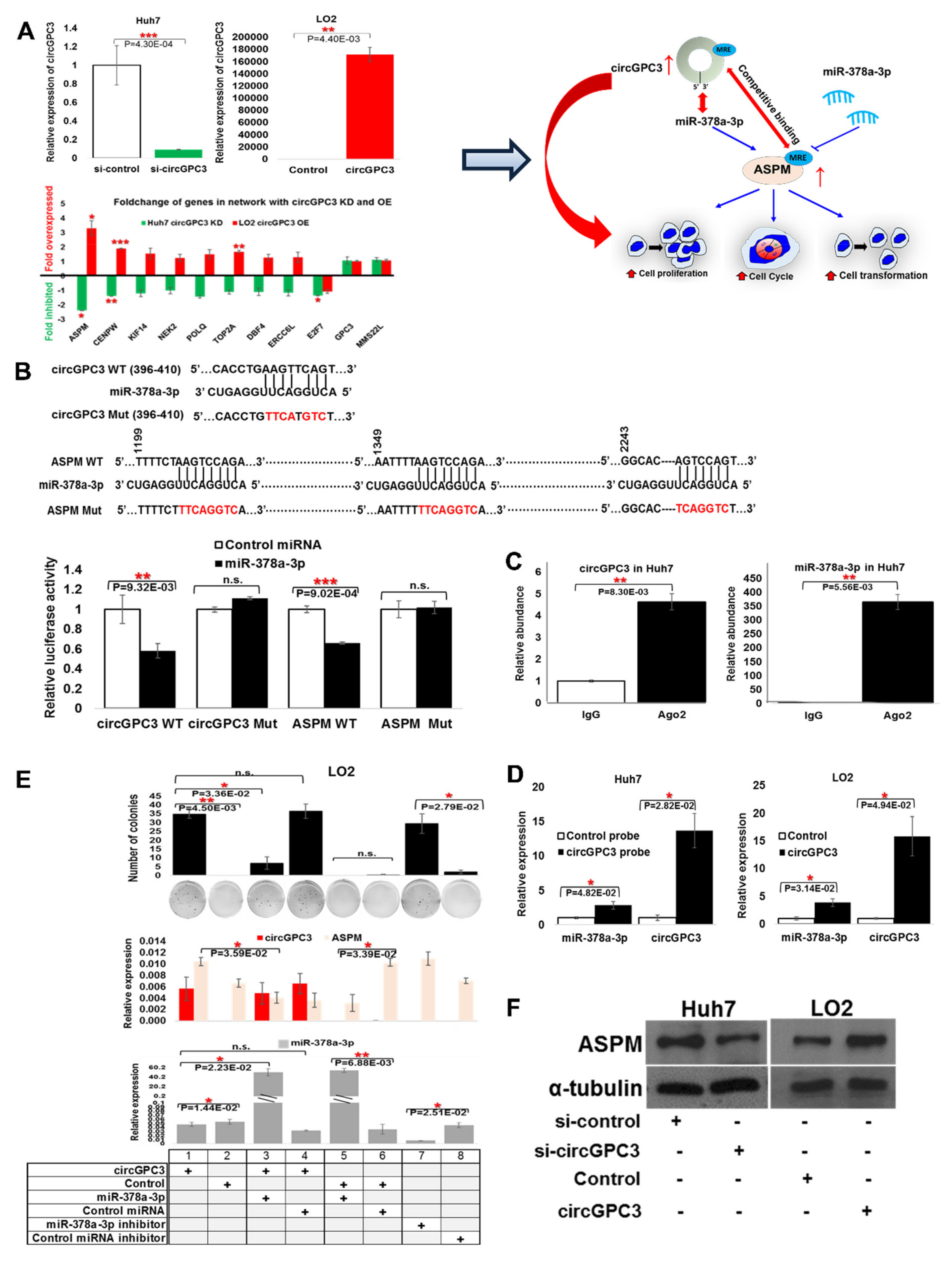
3.7. Role of circGPC3 in Modulating Tumorigenesis through Sponging miR-378a-3p to Upregulate ASPM
3.8. Significance of Clinically Relevant circRNA–Centric Regulatory Network in HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Matsuda, A.; Diederichs, S.; Patel, T. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J. Hepatol. 2017, 67, 603–618. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.J.; Wong, S.Y.S.; Huang, F.; Lim, S.; Chong, S.S.; Ooi, L.L.; Kon, O.L.; Lee, C.G. Roles and Regulation of Long Noncoding RNAs in Hepatocellular Carcinoma. Cancer Res. 2019, 79, 5131–5139. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, T.; Wang, X.; He, A. Circles reshaping the RNA world: From waste to treasure. Mol. Cancer 2017, 16, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Li, J.; Wang, H.; Su, X.; Hou, J.; Gu, Y.; Qian, C.; Lin, Y.; Liu, X.; Huang, M.; et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 2017, 66, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhao, G.; Yan, X.; Lv, Z.; Yin, H.; Zhang, S.; Song, W.; Li, X.; Li, L.; Du, Z.; et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018, 19, 218. [Google Scholar] [CrossRef] [Green Version]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37 e29. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Xu, Q.G.; Wang, Z.G.; Yang, Y.; Zhang, L.; Ma, J.Z.; Sun, S.H.; Yang, F.; Zhou, W.P. Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J. Hepatol. 2018, 68, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, X.; Zhang, N.; Chen, Z. Identification and integrated analysis of hepatocellular carcinoma-related circular RNA signature. Ann. Transl. Med. 2020, 8, 294. [Google Scholar] [CrossRef]
- Qiu, L.; Wang, T.; Ge, Q.; Xu, H.; Wu, Y.; Tang, Q.; Chen, K. Circular RNA Signature in Hepatocellular Carcinoma. J. Cancer 2019, 10, 3361–3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Cai, Y.; Xiong, H.; Deng, Y.; Dai, X. CCRDB: A cancer circRNAs-related database and its application in hepatocellular carcinoma-related circRNAs. Database 2019, 2019, baz063. [Google Scholar] [CrossRef]
- Deng, R.; Cui, X.; Dong, Y.; Tang, Y.; Tao, X.; Wang, S.; Wang, J.; Chen, L. Construction of circRNA-Based ceRNA Network to Reveal the Role of circRNAs in the Progression and Prognosis of Hepatocellular Carcinoma. Front. Genet. 2021, 12, 626764. [Google Scholar] [CrossRef]
- Xiong, D.D.; Dang, Y.W.; Lin, P.; Wen, D.Y.; He, R.Q.; Luo, D.Z.; Feng, Z.B.; Chen, G. A circRNA-miRNA-mRNA network identification for exploring underlying pathogenesis and therapy strategy of hepatocellular carcinoma. J. Transl. Med. 2018, 16, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, J.; Wang, W. Construction and investigation of breast-cancer-specific ceRNA network based on the mRNA and miRNA expression data. IET Syst. Biol. 2014, 8, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Y.; Lu, J.; Pan, T.; Ding, N.; Wang, Z.; Shao, T.; Zhang, J.; Wang, L.; Li, X. The mRNA related ceRNA-ceRNA landscape and significance across 20 major cancer types. Nucleic Acids Res. 2015, 43, 8169–8182. [Google Scholar] [CrossRef]
- Paci, P.; Colombo, T.; Farina, L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014, 8, 83. [Google Scholar] [CrossRef]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB—A database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011, 39, D712–D717. [Google Scholar] [CrossRef] [Green Version]
- Aufiero, S.; van den Hoogenhof, M.M.G.; Reckman, Y.J.; Beqqali, A.; van der Made, I.; Kluin, J.; Khan, M.A.F.; Pinto, Y.M.; Creemers, E.E. Cardiac circRNAs arise mainly from constitutive exons rather than alternatively spliced exons. RNA 2018, 24, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Iwakawa, H.O.; Tomari, Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef]
- Gao, W.; Ho, M. The role of glypican-3 in regulating Wnt in hepatocellular carcinomas. Cancer Rep. 2011, 1, 14–19. [Google Scholar] [PubMed]
- Gong, T.; Ning, X.; Deng, Z.; Liu, M.; Zhou, B.; Chen, X.; Huang, S.; Xu, Y.; Chen, Z.; Luo, R. Propofol-induced miR-219-5p inhibits growth and invasion of hepatocellular carcinoma through suppression of GPC3-mediated Wnt/beta-catenin signalling activation. J. Cell. Biochem. 2019, 120, 16934–16945. [Google Scholar] [CrossRef]
- Lin, S.Y.; Pan, H.W.; Liu, S.H.; Jeng, Y.M.; Hu, F.C.; Peng, S.Y.; Lai, P.L.; Hsu, H.C. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 4814–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, I.O.; Lai, E.C.; Ng, M.M.; Fan, S.T. Tumor encapsulation in hepatocellular carcinoma. A pathologic study of 189 cases. Cancer 1992, 70, 45–49. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, Y.K.; Byun, J.K.; Kim, M.K.; Kang, Y.N.; Kim, S.H.; Lee, S.; Jang, B.K.; Park, K.G. Estrogen-related receptor gamma is upregulated in liver cancer and its inhibition suppresses liver cancer cell proliferation via induction of p21 and p27. Exp. Mol. Med. 2016, 48, e213. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Han, S.; Peng, R.; Jiao, C.; Wang, X.; Yang, X.; Yang, R.; Li, X. Depletion of histone demethylase KDM5B inhibits cell proliferation of hepatocellular carcinoma by regulation of cell cycle checkpoint proteins p15 and p27. J. Exp. Clin. Cancer Res. 2016, 35, 37. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.L.; Zeng, Z.C.; Tang, Z.Y.; Fan, J.; Sun, H.C.; Tan, Y.S. Expression of cytokeratin 19 and matrix metalloproteinase 2 predicts lymph node metastasis in hepatocellular carcinoma. Mol. Biol. Rep. 2011, 38, 3531–3539. [Google Scholar] [CrossRef]
- Yu, G.R.; Kim, S.H.; Park, S.H.; Cui, X.D.; Xu, D.Y.; Yu, H.C.; Cho, B.H.; Yeom, Y.I.; Kim, S.S.; Kim, S.B.; et al. Identification of molecular markers for the oncogenic differentiation of hepatocellular carcinoma. Exp. Mol. Med. 2007, 39, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Feng, C.; Gao, M.; Jin, M.; Liu, T.; Yuan, Y.; Yan, G.; Gong, R.; Sun, Y.; He, M.; et al. MicroRNA-92b-5p modulates melatonin-mediated osteogenic differentiation of bone marrow mesenchymal stem cells by targeting ICAM-1. J. Cell. Mol. Med. 2019, 23, 6140–6153. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Zeng, M.; Zhu, L.; Li, L.; Kang, C. miR-378 suppresses the proliferation, migration and invasion of colon cancer cells by inhibiting SDAD1. Cell. Mol. Biol. Lett. 2017, 22, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikeye, S.N.; Colin, C.; Marie, Y.; Vampouille, R.; Ravassard, P.; Rousseau, A.; Boisselier, B.; Idbaih, A.; Calvo, C.F.; Leuraud, P.; et al. Correction: ASPM-associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell. Int. 2011, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Capecchi, M.R.; Pozner, A. ASPM regulates symmetric stem cell division by tuning Cyclin E ubiquitination. Nat. Commun. 2015, 6, 8763. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; He, H.; Zhang, Y.; Wang, X.; Yang, L.; An, Z. CCNB2, TOP2A, and ASPM Reflect the Prognosis of Hepatocellular Carcinoma, as Determined by Weighted Gene Coexpression Network Analysis. Biomed. Res. Int. 2020, 2020, 4612158. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lu, M.; Cui, Q.; Zhang, D.; Kong, D.; Liao, X.; Ren, J.; Gong, Y.; Wu, G. Overexpression of ASPM, CDC20, and TTK Confer a Poorer Prognosis in Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2019, 9, 310. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Veno, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfafenrot, C.; Preusser, C. Establishing essential quality criteria for the validation of circular RNAs as biomarkers. Biomol. Detect. Quantif. 2019, 17, 100085. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Chen, S.; Cao, Q.; Wen, W.; Wang, H. Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett. 2019, 460, 1–9. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, M.; Ma, H.; Song, X.; He, L.; Ye, X.; Li, X. Overexpression of glypican-3 is a predictor of poor prognosis in hepatocellular carcinoma: An updated meta-analysis. Medicine 2018, 97, e11130. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Fujinami, N.; Shimizu, Y.; Mizuno, S.; Saito, K.; Suzuki, T.; Konishi, M.; Takahashi, S.; Gotohda, N.; Suto, K.; et al. Usefulness of plasma full-length glypican-3 as a predictive marker of hepatocellular carcinoma recurrence after radial surgery. Oncol. Lett. 2020, 19, 2657–2666. [Google Scholar] [CrossRef]
- Nishida, T.; Kataoka, H. Glypican 3-Targeted Therapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Thomson Brooks/Cole Publishing Co.: Pacific Grove, CA, USA, 1996. [Google Scholar]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2018, 58, 588–602. [Google Scholar] [CrossRef]
- Costa, P.; Gautrot, J.E.; Connelly, J.T. Directing cell migration using micropatterned and dynamically adhesive polymer brushes. Acta Biomater. 2014, 10, 2415–2422. [Google Scholar] [CrossRef]
- Su, X.; Wang, H.; Ge, W.; Yang, M.; Hou, J.; Chen, T.; Li, N.; Cao, X. An In Vivo Method to Identify microRNA Targets Not Predicted by Computation Algorithms: p21 Targeting by miR-92a in Cancer. Cancer Res. 2015, 75, 2875–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.V.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2010, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Thurnherr, T.; Chung, A.Y.F.; Goh, B.K.P.; Chow, P.K.H.; Chan, C.Y.; Cheow, P.C.; Lee, S.Y.; Lim, T.K.H.; Chong, S.S.; et al. Clinicopathological-Associated Regulatory Network of Deregulated circRNAs in Hepatocellular Carcinoma. Cancers 2021, 13, 2772. https://doi.org/10.3390/cancers13112772
Han J, Thurnherr T, Chung AYF, Goh BKP, Chow PKH, Chan CY, Cheow PC, Lee SY, Lim TKH, Chong SS, et al. Clinicopathological-Associated Regulatory Network of Deregulated circRNAs in Hepatocellular Carcinoma. Cancers. 2021; 13(11):2772. https://doi.org/10.3390/cancers13112772
Chicago/Turabian StyleHan, Jian, Thomas Thurnherr, Alexander Y. F. Chung, Brian K. P. Goh, Pierce K. H. Chow, Chung Yip Chan, Peng Chung Cheow, Ser Yee Lee, Tony K. H. Lim, Samuel S. Chong, and et al. 2021. "Clinicopathological-Associated Regulatory Network of Deregulated circRNAs in Hepatocellular Carcinoma" Cancers 13, no. 11: 2772. https://doi.org/10.3390/cancers13112772