
Driving CAR T Stem Cell Targeting in Acute Myeloid Leukemia: The Roads to Success



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Role of LSC and the BM Microenvironment in AML
2.1. LSC vs. HSC: Selective LSC-Targeted Eradication
2.2. Sources of Heterogeneity in AML-LSC
2.3. The Role of the BM Niche in AML
3. CAR T-Cells and Their Current Clinical Application in AML Therapy
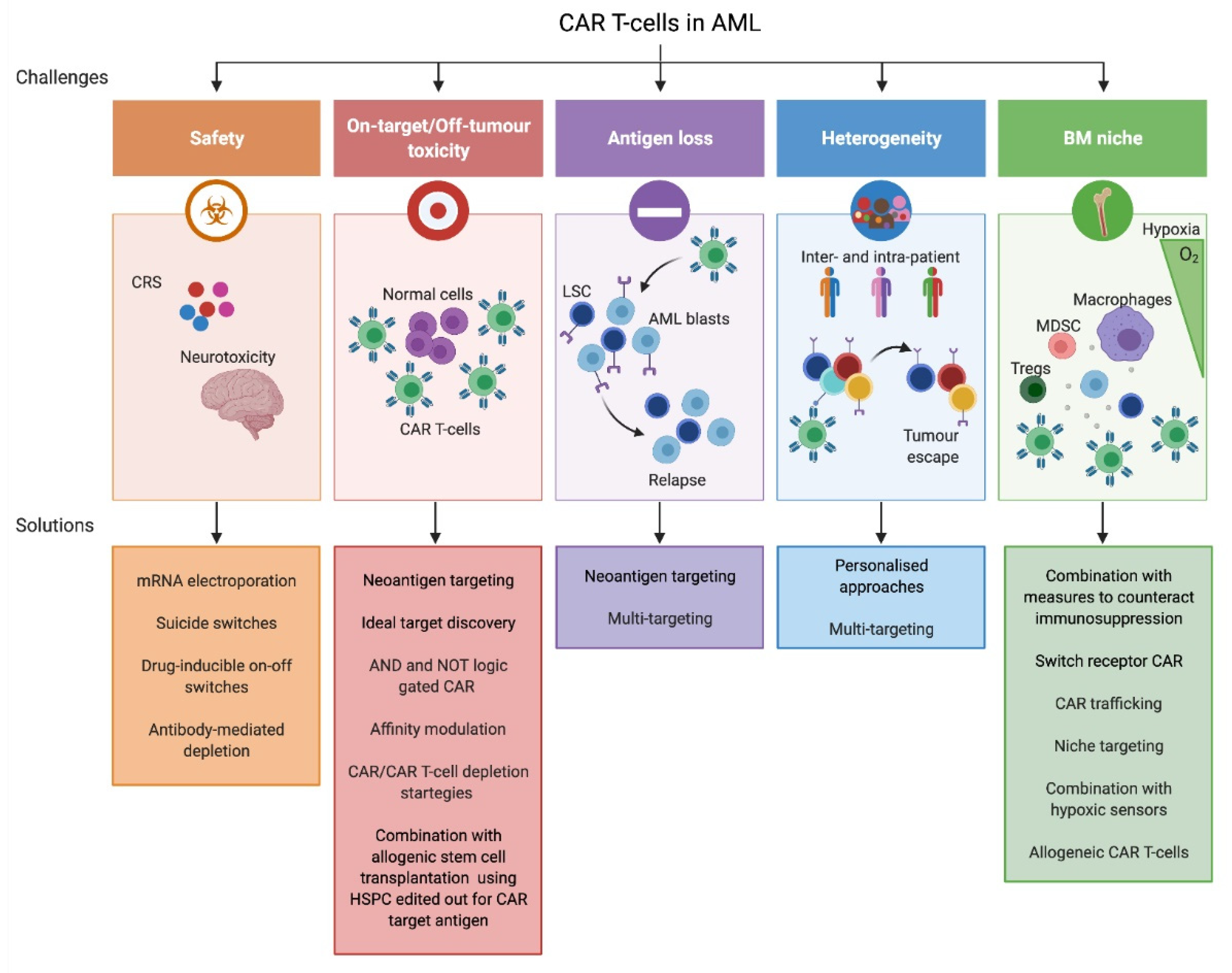
4. CAR T-Cell Approaches in AML: Challenges and Novel Strategies
4.1. CAR T-Cell Antigens in AML and Strategies to Overcome on-Target/Off-Tumour Effects
4.2. AML Phenotypic Heterogeneity Challenges the Identification of Target Antigens for AML CAR T-Cell Strategies
4.3. Targeting the AML Tumour Microenvironment with CAR T-Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- American Cancer Society. Key Statistics for Acute Myeloid Leukemia (AML). Available online: https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html (accessed on 9 May 2021).
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, E.N.; Franek, J.; Patel, D.; Zaidi, O.; Nehme, S.A.; Almeida, A.M. The Real-World Incidence of Relapse in Acute Myeloid Leukemia (AML): A Systematic Literature Review (SLR). Blood 2018, 132, 5188. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Uddin, S.; Mohammad, R.M. Targeting acute myeloid leukemia stem cell signaling by natural products. Mol. Cancer 2017, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, M.; Abe, A.; Katsumi, A.; Xu, J.; Ito, M.; Arai, F.; Suda, T.; Ito, M.; Kiyoi, H.; Kinoshita, T.; et al. Homing, proliferation and survival sites of human leukemia cells in vivo in immunodeficient mice. Leukemia 2007, 21, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Korn, C.; Mendez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, A.S.; Kim, K.T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882. [Google Scholar] [CrossRef]
- Stahl, M.; Lu, B.Y.; Kim, T.K.; Zeidan, A.M. Novel Therapies for Acute Myeloid Leukemia: Are We Finally Breaking the Deadlock? Target. Oncol. 2017, 12, 413–447. [Google Scholar] [CrossRef]
- Tan, Y.; Wu, Q.; Zhou, F. Targeting acute myeloid leukemia stem cells: Current therapies in development and potential strategies with new dimensions. Crit. Rev. Oncol. Hematol. 2020, 152, 102993. [Google Scholar] [CrossRef]
- Karantanou, C.; Godavarthy, P.S.; Krause, D.S. Targeting the bone marrow microenvironment in acute leukemia. Leuk. Lymphoma 2018, 59, 2535–2545. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.M.; Dobson, S.M.; Voisin, V.; McLeod, J.; Kennedy, J.A.; Mitchell, A.; Jin, L.; Eppert, K.; Bader, G.; Minden, M.D.; et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020, 4, 5402–5413. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.J. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J. Stem Cells 2014, 6, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Zhao, S.; Hu, W.; Jiang, S.; Snell, V.; Weidner, D.; Jackson, C.E.; Zhang, X.; Champlin, R.; Estey, E.; et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br. J. Haematol. 2002, 118, 521–534. [Google Scholar] [CrossRef]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy. Crit. Rev. Oncol. Hematol. 2019, 144, 102814. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelozzi, I.M.; Granata, V.; De Ponti, G.; Alberti, G.; Tomasoni, C.; Antolini, L.; Gambacorti-Passerini, C.; Gentner, B.; Dazzi, F.; Biondi, A.; et al. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.J.; Doondeea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J. Exp. Med. 2016, 213, 1513–1535. [Google Scholar] [CrossRef]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Lowenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef] [Green Version]
- Van Rhenen, A.; Feller, N.; Kelder, A.; Westra, A.H.; Rombouts, E.; Zweegman, S.; van der Pol, M.A.; Waisfisz, Q.; Ossenkoppele, G.J.; Schuurhuis, G.J. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin. Cancer Res. 2005, 11, 6520–6527. [Google Scholar] [CrossRef] [Green Version]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Behbehani, G.K.; Samusik, N.; Bjornson, Z.B.; Fantl, W.J.; Medeiros, B.C.; Nolan, G.P. Mass Cytometric Functional Profiling of Acute Myeloid Leukemia Defines Cell-Cycle and Immunophenotypic Properties That Correlate with Known Responses to Therapy. Cancer Discov. 2015, 5, 988–1003. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Rulicke, T.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi Najafabadi, M.; Shamsasenjan, K.; Akbarzadehalaleh, P. Angiogenesis Status in Patients with Acute Myeloid Leukemia: From Diagnosis to Post-hematopoietic Stem Cell Transplantation. Int. J. Organ. Transplant. Med. 2017, 8, 57–67. [Google Scholar]
- Chen, C.; Liu, Y.; Hua, M.; Li, X.; Ji, C.; Ma, D. Neuropathy correlated with imbalanced Foxp3/IL-17 in bone marrow microenvironment of patients with acute myeloid leukemia. Oncotarget 2016, 7, 24455–24465. [Google Scholar] [CrossRef]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.L.; Reid, J.C.; Salci, K.R.; Aslostovar, L.; Benoit, Y.D.; Shapovalova, Z.; Nakanishi, M.; Porras, D.P.; Almakadi, M.; Campbell, C.J.V.; et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat. Cell Biol. 2017, 19, 1336–1347. [Google Scholar] [CrossRef]
- Bernasconi, P.; Borsani, O. Targeting Leukemia Stem Cell-Niche Dynamics: A New Challenge in AML Treatment. J. Oncol. 2019, 2019, 8323592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkaliaris, K.D.; Scadden, D.T. Cell interactions in the bone marrow microenvironment affecting myeloid malignancies. Blood Adv. 2020, 4, 3795–3803. [Google Scholar] [CrossRef]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932 e1916. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Salvestrini, V.; Ciciarello, M.; Loscocco, F.; Visani, G.; Parisi, S.; Lecciso, M.; Ocadlikova, D.; Rossi, L.; Gabucci, E.; et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev. Hematol. 2014, 7, 807–818. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, H.; Pepper, A.; Chevassut, T. Acute Myeloid Leukaemia in Its Niche: The Bone Marrow Microenvironment in Acute Myeloid Leukaemia. Curr. Oncol. Rep. 2020, 22, 27. [Google Scholar] [CrossRef] [Green Version]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Theocharides, A.P.; Jin, L.; Cheng, P.Y.; Prasolava, T.K.; Malko, A.V.; Ho, J.M.; Poeppl, A.G.; van Rooijen, N.; Minden, M.D.; Danska, J.S.; et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 2012, 209, 1883–1899. [Google Scholar] [CrossRef] [Green Version]
- Villatoro, A.; Konieczny, J.; Cuminetti, V.; Arranz, L. Leukemia Stem Cell Release from the Stem Cell Niche to Treat Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 607. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Bruserud, O.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar] [PubMed]
- Zhai, Y.; Zhang, J.; Wang, H.; Lu, W.; Liu, S.; Yu, Y.; Weng, W.; Ding, Z.; Zhu, Q.; Shi, J. Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J. Exp. Clin. Cancer Res. 2016, 35, 147. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.W.; Shi, J.; Chen, J.; Wang, B.; Yu, Y.H.; Qin, X.; Zhou, X.C.; Cai, Y.J.; Li, Z.Q.; Zhang, F.; et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 2014, 25, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takam Kamga, P.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Imataki, O.; Torii, E.; Kameda, T.; Shide, K.; Shimoda, H.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Yamamoto, S.; et al. Elevated HIF-1alpha expression of acute myelogenous leukemia stem cells in the endosteal hypoxic zone may be a cause of minimal residual disease in bone marrow after chemotherapy. Leuk. Res. 2012, 36, e122–e124. [Google Scholar] [CrossRef]
- Drolle, H.; Wagner, M.; Vasold, J.; Kutt, A.; Deniffel, C.; Sotlar, K.; Sironi, S.; Herold, T.; Rieger, C.; Fiegl, M. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk. Res. 2015, 39, 779–785. [Google Scholar] [CrossRef]
- Bray, L.J.; Binner, M.; Korner, Y.; von Bonin, M.; Bornhauser, M.; Werner, C. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica 2017, 102, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Honemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Fiorenza, S.; Turtle, C.J. CAR-T Cell Therapy for Acute Myeloid Leukemia: Preclinical Rationale, Current Clinical Progress, and Barriers to Success. BioDrugs 2021, 35, 281–302. [Google Scholar] [CrossRef] [PubMed]
- Bu, C.; Peng, Z.; Luo, M.; Li, G.; Li, C. Phase I Clinical Trial of Autologous CLL1 CAR-T Therapy for Pediatric Patients with Relapsed and Refractory Acute Myeloid Leukemia. Blood 2020, 136, 13. [Google Scholar] [CrossRef]
- Chen, L.; Mao, H.; Zhang, J.; Chu, J.; Devine, S.; Caligiuri, M.A.; Yu, J. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia 2017, 31, 1830–1834. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Yoshida, T.; Mihara, K.; Takei, Y.; Yanagihara, K.; Kubo, T.; Bhattacharyya, J.; Imai, C.; Mino, T.; Takihara, Y.; Ichinohe, T. All-trans retinoic acid enhances cytotoxic effect of T cells with an anti-CD38 chimeric antigen receptor in acute myeloid leukemia. Clin. Transl. Immunol. 2016, 5, e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Poussin, M.; Kalota, A.; Feng, Y.; Low, P.S.; Dimitrov, D.S.; Powell, D.J., Jr. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 2015, 125, 3466–3476. [Google Scholar] [CrossRef]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J., Jr. High-affinity FRbeta-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef]
- Rafiq, S.; Purdon, T.J.; Daniyan, A.F.; Koneru, M.; Dao, T.; Liu, C.; Scheinberg, D.A.; Brentjens, R.J. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2017, 31, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, N.; Midorikawa, A.; Ida, K.; Fudaba, Y.W.; Isobe, M. Development of a T-cell receptor mimic antibody targeting a novel Wilms tumor 1-derived peptide and analysis of its specificity. Cancer Sci. 2020, 111, 3516–3526. [Google Scholar] [CrossRef]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3–specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, C.; Liu, Z.; Yang, M.; Tang, X.; Wang, Y.; Zheng, M.; Huang, J.; Zhong, K.; Zhao, S.; et al. B7-H3-Targeted CAR-T Cells Exhibit Potent Antitumor Effects on Hematologic and Solid Tumors. Mol. Ther. Oncolytics 2020, 17, 180–189. [Google Scholar] [CrossRef]
- Leick, M.; Scarfò, I.; Choi, B.D.; Larson, R.; Bouffard, A.A.; Castano, A.; Cabral, M.L.; Schmidts, A.; Frigault, M.J.; Maus, M.V. Use of CD70 Targeted Chimeric Antigen Receptor (CAR) T Cells for the Treatment of Acute Myeloid Leukemia (AML). Blood 2019, 134, 4443. [Google Scholar] [CrossRef]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.N.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Muller-Tidow, C.; et al. CD70-specific CAR T-cells have potent activity against Acute Myeloid Leukemia (AML) without HSC toxicity. Blood 2021. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.S.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: How far up the road have we traveled? Ther. Adv. Hematol. 2018, 9, 135–148. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrot, S.; Hallam, S. CAR-T Cells: Future Perspectives. Hemasphere 2019, 3, e188. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Xie, G.; Ivica, N.A.; Jia, B.; Li, Y.; Dong, H.; Liang, Y.; Brown, D.; Rizwan, R.; Chen, J. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng. 2020. [Google Scholar] [CrossRef]
- Graf, C.; Heidel, F.; Tenzer, S.; Radsak, M.P.; Solem, F.K.; Britten, C.M.; Huber, C.; Fischer, T.; Wolfel, T. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 2007, 109, 2985–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, M.; Hourigan, C.S. Novel Antigen Targets for Immunotherapy of Acute Myeloid Leukemia. Curr. Drug Targets 2017, 18, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Depreter, B.; Weening, K.E.; Vandepoele, K.; Essand, M.; De Moerloose, B.; Themeli, M.; Cloos, J.; Hanekamp, D.; Moors, I.; D’Hont, I.; et al. TARP is an immunotherapeutic target in acute myeloid leukemia expressed in the leukemic stem cell compartment. Haematologica 2020, 105, 1306–1316. [Google Scholar] [CrossRef]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubner, S.; Perna, F.; Kohnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Petrov, J.C.; Wada, M.; Pinz, K.G.; Yan, L.E.; Chen, K.H.; Shuai, X.; Liu, H.; Chen, X.; Leung, L.H.; Salman, H.; et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia 2018, 32, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H.; et al. Treating Relapsed / Refractory (RR) AML with Biodegradable Anti-CD123 CAR Modified T Cells. Blood 2017, 130, 1359. [Google Scholar] [CrossRef]
- Willier, S.; Rothamel, P.; Hastreiter, M.; Wilhelm, J.; Stenger, D.; Blaeschke, F.; Rohlfs, M.; Kaeuferle, T.; Schmid, I.; Albert, M.H.; et al. CLEC12A and CD33 co-expression as preferential target on pediatric AML for combinatorial immunotherapy. Blood 2021, 137, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Danylesko, I.; Jacoby, E.; Yerushalmi, R.; Shem-Tov, N.; Besser, M.J.; Vernitsky, H.; Marcu-Malina, V.; Shimoni, A.; Avigdor, A.; Nagler, A. Remission of acute myeloid leukemia with t(8;21) following CD19 CAR T-cells. Leukemia 2020, 34, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhang, H.; Sun, L.; Li, Y.; Zhang, S.; He, G.; Yi, H.; Wada, M.; Pinz, K.G.; Chen, K.H.; et al. First-in-human CLL1-CD33 compound CAR (cCAR) T cell therapy in relapsed and refractory acute myeloid leukemia. In Proceedings of the 25th EHA Annual Congress, Frankfurt, Germany, 12 June 2020. [Google Scholar]
- Epperly, R.; Gottschalk, S.; Velasquez, M.P. A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front. Oncol. 2020, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, X.; Zhang, H.; Liang, Z.; Shao, M.; Wang, X.; Ding, L.; Cui, J.; Hu, Y.; Huang, H. Galectin-9 from Bone Marrow Mesenchymal Stromal Cells Mediates Immunosuppression on Chimeric Antigen Receptor T Cells. Blood 2019, 134, 5609. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti, S.R.; Romecin, P.A.; Vinyoles, M.; Juan, M.; Fuster, J.L.; Camos, M.; Querol, S.; Delgado, M.; Menendez, P. Bone marrow MSC from pediatric patients with B-ALL highly immunosuppress T-cell responses but do not compromise CD19-CAR T-cell activity. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Choi, U.; Corsino, C.I.; Koontz, S.M.; Tajima, M.; Sweeney, C.L.; Black, M.A.; Feldman, S.A.; Dinauer, M.C.; Malech, H.L. Myeloid Conditioning with c-kit-Targeted CAR-T Cells Enables Donor Stem Cell Engraftment. Mol. Ther. 2018, 26, 1181–1197. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, C.W.; Urak, R.; Taus, E.; Aguilar, B.; Chang, W.C.; Mardiros, A.; Budde, L.E.; Brown, C.E.; Berger, C.; et al. Comparison of naive and central memory derived CD8(+) effector cell engraftment fitness and function following adoptive transfer. Oncoimmunology 2016, 5, e1072671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sironi, S.; Wagner, M.; Kuett, A.; Drolle, H.; Polzer, H.; Spiekermann, K.; Rieger, C.; Fiegl, M. Microenvironmental hypoxia regulates FLT3 expression and biology in AML. Sci. Rep. 2015, 5, 17550. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Cheng, H.Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelozzi, I.M.; Kirtsios, E.; Giustacchini, A. Driving CAR T Stem Cell Targeting in Acute Myeloid Leukemia: The Roads to Success. Cancers 2021, 13, 2816. https://doi.org/10.3390/cancers13112816
Michelozzi IM, Kirtsios E, Giustacchini A. Driving CAR T Stem Cell Targeting in Acute Myeloid Leukemia: The Roads to Success. Cancers. 2021; 13(11):2816. https://doi.org/10.3390/cancers13112816
Chicago/Turabian StyleMichelozzi, Ilaria M., Efstratios Kirtsios, and Alice Giustacchini. 2021. "Driving CAR T Stem Cell Targeting in Acute Myeloid Leukemia: The Roads to Success" Cancers 13, no. 11: 2816. https://doi.org/10.3390/cancers13112816
APA StyleMichelozzi, I. M., Kirtsios, E., & Giustacchini, A. (2021). Driving CAR T Stem Cell Targeting in Acute Myeloid Leukemia: The Roads to Success. Cancers, 13(11), 2816. https://doi.org/10.3390/cancers13112816