
Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Neu/HER2 in Breast Cancer
2.1. Neu/HER2 Biology
2.2. HER2 Inhibitors: Mechanisms of Action and Efficacy in Clinical Studies
2.2.1. Trastuzumab (Herceptin®)
2.2.2. Lapatinib
2.2.3. Neratinib
2.2.4. Pertuzumab (2C4)
2.2.5. Trastuzumab Antibody–Drug Conjugates
2.2.6. Tucatinib
2.2.7. Pyrotinib
2.2.8. HLX02
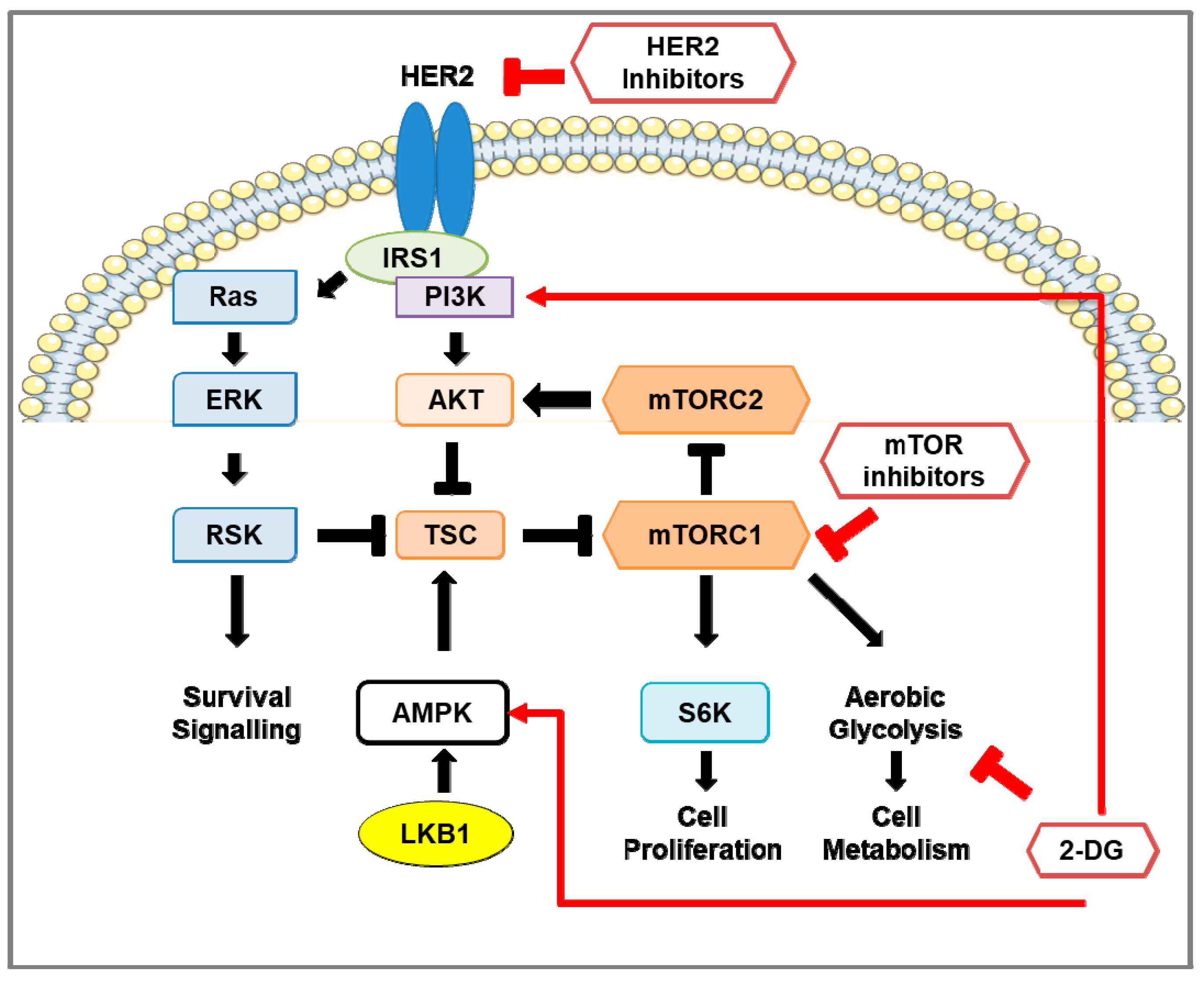
3. The mTOR Pathway in HER2-Positive Breast Cancer
3.1. mTOR Protein Structure and Function
3.2. Regulation of mTOR Pathway
3.3. Downstream of mTOR Pathway
3.3.1. mTORC1 Activation of S6K1 and 4E-BP1
3.3.2. mTORC1 Regulation of Cell Cycle
3.3.3. mTORC1 Regulation of Akt
3.3.4. mTORC1 Regulation of Glycolysis
3.3.5. mTORC1 Regulation of Mitochondrial Biogenesis
3.3.6. mTORC1 Regulation of Autophagy
3.3.7. mTORC1 Regulation of mTORC2
3.3.8. Downstream of mTORC2
3.3.9. mTORC2 Regulation of Glycolysis
3.3.10. Role of mTORC1 and mTORC2 in Immunity
3.4. mTOR Pathway Inhibitors: Mechanisms of Action and Efficacy in Clinical Studies for the Treatment of HER2-Positive Breast Cancer
3.4.1. Rapamycin (Sirolimus) and Rapalogs
3.4.2. mTOR Kinase Inhibitors (TKIs)
3.4.3. Dual PI3K-mTOR Inhibitors
3.5. Clinical Response of mTOR Inhibition in HER2-Positive Breast Cancer Patients

{kind=link}
{kind=link}
{kind=link}
| Treatment | Trial Name | Patients | ORR | Median PFS | OS (Months) | REF | |
|---|---|---|---|---|---|---|---|
| Everolimus | EV, V, T | NCT0046556 Phase Ib/II | 33 | 44.00% | 34 weeks | n/a | [164] |
| EV, EX | NCT00426530 Phase Ib | 50 | 19.10% | 30.7 weeks | n/a | [176] | |
| EV, P, T | BOLERO-1 (NCT00876395) Phase III | 719 | 67.00% | 14.95 months | 63% survival at 41.3 | [165] | |
| 303 (Asian subset) | 69.20% | 18.40 months, 25.46 months (HR-) | n/a | [166] | |||
| EV, P, T | BOLERO-3 (NCT01007942) Phase III | 569 | 41.00% | 7 months | n/a | [167] | |
| EV, P, T | LCCC 1025 (NCT01305941) Phase II | 32 | CBR: 20% (>6 months), IRR: 4% | 3.93 months (TTP) | 12.2 | [169] | |
| Ridaforolimus | R, T | NCT00736970 Phase IIb | 34 | CBR: 34.3% | 5.4 months | 17.7 | [168] |
| Dactolisib (BEZ235) | B | NCT00620594 PhaseI/Ib | 153 | 0% | SD: 31.1–42.4% | n/a | [173] |
| B, T | 30 | 13.30% | SD: 40% | n/a | |||
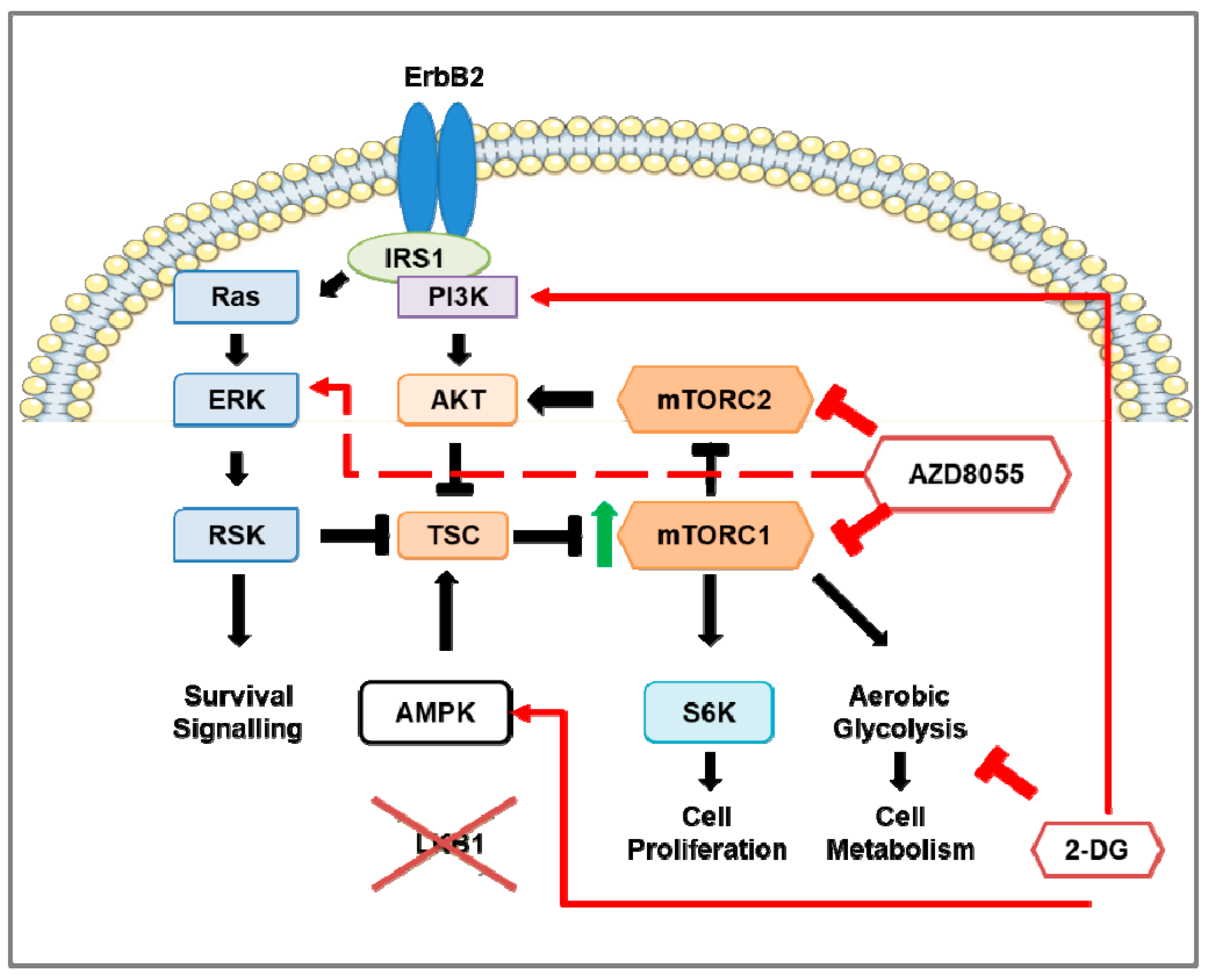
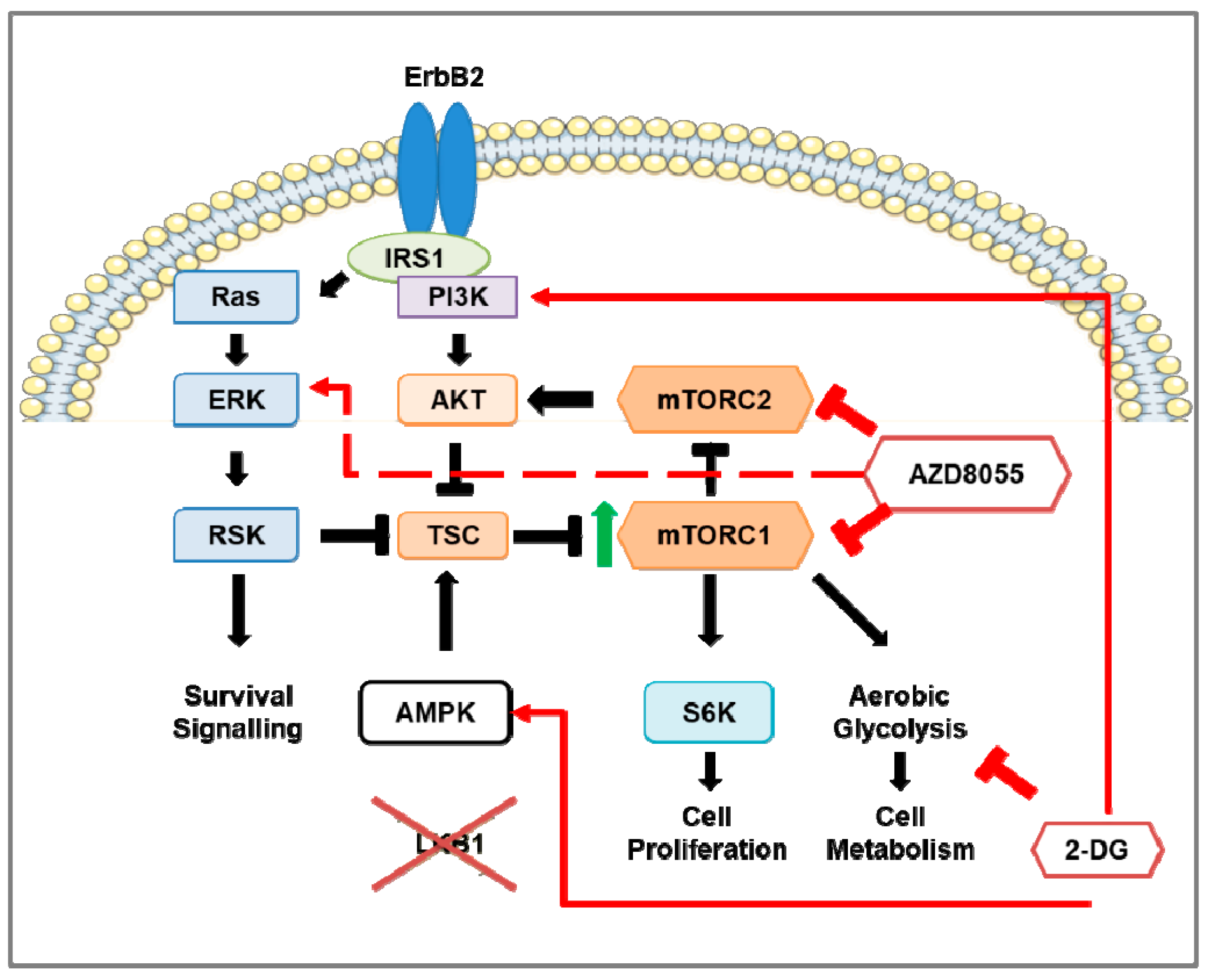
3.6. Preclinical Studies of mTOR Pathway in HER2-Positive Breast Cancer Mouse Models
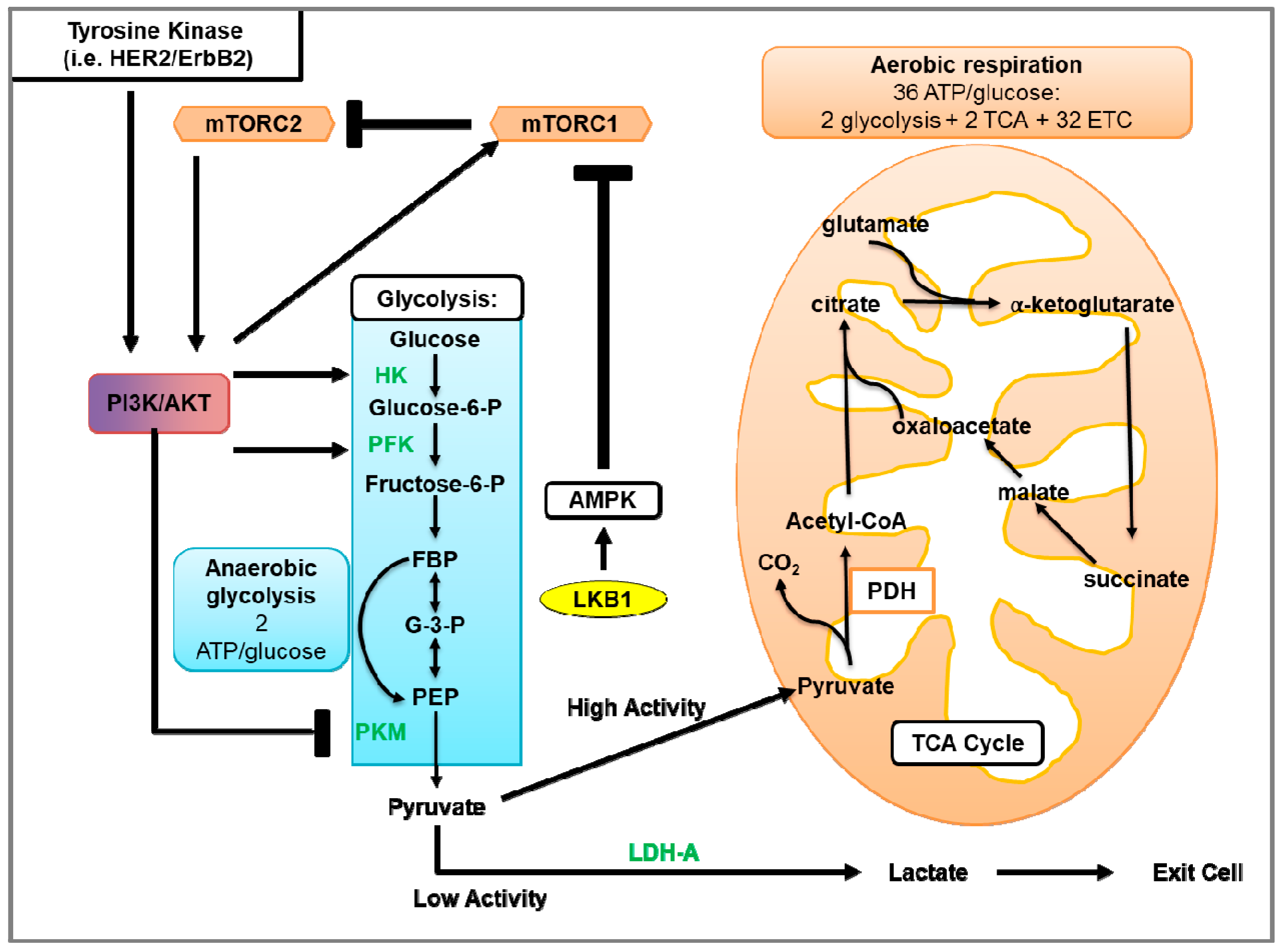
4. Glycolysis in Breast Cancer
4.1. Glycolysis and Oxidative Phosphorylation
4.2. Glycolysis in Cancer
4.3. Glycolysis in HER2-Positive Breast Cancer and the Involvement of mTOR
4.4. Glucose Analogs and the Effect on Cancer Cells
4.5. Adverse Effects of 2-DG
4.6. Combination Therapy of mTOR Inhibition and 2-DG in HER2-Positive Breast Cancer Mouse Models
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Ding, P.; Wu, Y.; Tian, W.; Pan, T.; Zhang, S. Global patterns and trends in the breast cancer incidence and mortality according to sociodemographic indices: An observational study based on the global burden of diseases. BMJ Open 2019, 9, e028461. [Google Scholar] [CrossRef]
- Chatenoud, L.; Bertuccio, P.; Bosetti, C.; Malvezzi, M.; Levi, F.; Negri, E.; La Vecchia, C. Trends in mortality from major cancers in the Americas: 1980–2010. Ann.Oncol. 2014, 25, 1843–1853. [Google Scholar] [CrossRef]
- Anderson, B.O.; Cazap, E.; El Saghir, N.S.; Yip, C.-H.; Khaled, H.M.; Otero, I.V.; Adebamowo, C.A.; Badwe, R.A.; Harford, J.B. Optimisation of breast cancer management in low-resource and middle-resource countries: Executive summary of the Breast Health Global Initiative consensus, 2010. Lancet Oncol. 2011, 12, 387–398. [Google Scholar] [CrossRef]
- Li, N.; Deng, Y.; Zhou, L.; Tian, T.; Yang, S.; Wu, Y.; Zheng, Y.; Zhai, Z.; Hao, Q.; Song, D.; et al. Global burden of breast cancer and attributable risk factors in 195 countries and territories, from 1990 to 2017: Results from the Global Burden of Disease Study 2017. J. Hematol. Oncol. 2019, 12, 140. [Google Scholar] [CrossRef]
- Moasser, M.M.; Krop, I.E. The Evolving Landscape of HER2 Targeting in Breast Cancer. JAMA Oncol. 2015, 1, 1154–1161. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol.J. 1996, 16, 5276–5287. [Google Scholar] [CrossRef] [Green Version]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Llombart-Cussac, A.; Cortés, J.; Paré, L.; Galván, P.; Bermejo, B.; Martínez, N.; Vidal, M.; Pernas, S.; López, R.; Muñoz, M.; et al. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): An open-label, single-group, multicentre, phase 2 trial. Lancet Oncol. 2017, 18, 545–554. [Google Scholar] [CrossRef]
- Prat, A.; Pascual, T.; Adamo, B. Intrinsic molecular subtypes of HER2+ breast cancer. Oncotarget 2017, 8, 73362–73363. [Google Scholar] [CrossRef]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef] [Green Version]
- Järvinen, T.A.; Kononen, J.; Pelto-Huikko, M.; Isola, J. Expression of topoisomerase IIalpha is associated with rapid cell proliferation, aneuploidy, and c-erbB2 overexpression in breast cancer. Am. J. Pathol. 1996, 148, 2073–2082. [Google Scholar]
- Ross, J.S.; Fletcher, J.A. The HER-2/neu Oncogene in Breast Cancer: Prognostic Factor, Predictive Factor, and Target for Therapy. STEM CELLS 1998, 16, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, M.; ten Klooster, J.P.; Offerhaus, G.J.; Clevers, H. LKB1 and AMPK Family Signaling: The Intimate Link Between Cell Polarity and Energy Metabolism. Physiol. Rev. 2009, 89, 777–798. [Google Scholar] [CrossRef] [Green Version]
- Klapper, L.N.; Waterman, H.; Sela, M.; Yarden, Y. Tumor-inhibitory Antibodies to HER-2/ErbB-2 May Act by Recruiting c-Cbl and Enhancing Ubiquitination of HER-2. Cancer Res. 2000, 60, 3384. [Google Scholar]
- FDA. Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=reportsSearch.process (accessed on 1 March 2021).
- Maadi, H.; Nami, B.; Tong, J.; Li, G.; Wang, Z. The effects of trastuzumab on HER2-mediated cell signaling in CHO cells expressing human HER2. BMC Cancer 2018, 18, 238. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Klapper, L.N.; Vaisman, N.; Hurwitz, E.; Pinkas-Kramarski, R.; Yarden, Y.; Sela, M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene 1997, 14, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Tripathy, D.; Mendelsohn, J.; Baughman, S.; Benz, C.C.; Dantis, L.; Sklarin, N.T.; Seidman, A.D.; Hudis, C.A.; Moore, J.; et al. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Semin. Oncol. 1999, 26, 78–83. [Google Scholar] [PubMed]
- Pegram, M.D.; Pienkowski, T.; Northfelt, D.W.; Eiermann, W.; Patel, R.; Fumoleau, P.; Quan, E.; Crown, J.; Toppmeyer, D.; Smylie, M.; et al. Results of Two Open-Label, Multicenter Phase II Studies of Docetaxel, Platinum Salts, and Trastuzumab in HER2-Positive Advanced Breast Cancer. JNCI J. Natl. Cancer Inst. 2004, 96, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Shao, B.; Yan, Y.; Song, G.; Liu, X.; Wang, J.; Liang, X. Efficacy and safety of trastuzumab combined with chemotherapy for first-line treatment and beyond progression of HER2-overexpressing advanced breast cancer. Chin. J. Cancer Res. 2016, 28, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumsri, S.; Li, Z.; Serie, D.J.; Mashadi-Hossein, A.; Colon-Otero, G.; Song, N.; Pogue-Geile, K.L.; Gavin, P.G.; Paik, S.; Moreno-Aspitia, A.; et al. Incidence of Late Relapses in Patients With HER2-Positive Breast Cancer Receiving Adjuvant Trastuzumab: Combined Analysis of NCCTG N9831 (Alliance) and NRG Oncology/NSABP B-31. J. Clin. Oncol. 2019, 37, 3425–3435. [Google Scholar] [CrossRef]
- Baker, J.H.E.; Kyle, A.H.; Reinsberg, S.A.; Moosvi, F.; Patrick, H.M.; Cran, J.; Saatchi, K.; Häfeli, U.; Minchinton, A.I. Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: Role of the tumor microenvironment. Clin. Exp. Metastasis 2018, 35, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Seidman, A.; Hudis, C.; Pierri, M.K.; Shak, S.; Paton, V.; Ashby, M.; Murphy, M.; Stewart, S.J.; Keefe, D. Cardiac Dysfunction in the Trastuzumab Clinical Trials Experience. J. Clin. Oncol. 2002, 20, 1215–1221. [Google Scholar] [CrossRef]
- Crone, S.A.; Zhao, Y.-Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef]
- Ozcelik, C.; Erdmann, B.; Pilz, B.; Wettschureck, N.; Britsch, S.; Hübner, N.; Chien, K.R.; Birchmeier, C.; Garratt, A.N. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 8880–8885. [Google Scholar] [CrossRef] [Green Version]
- Gomez, H.L.; Doval, D.C.; Chavez, M.A.; Ang, P.C.S.; Aziz, Z.; Nag, S.; Ng, C.; Franco, S.X.; Chow, L.W.C.; Arbushites, M.C.; et al. Efficacy and Safety of Lapatinib As First-Line Therapy for ErbB2-Amplified Locally Advanced or Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 2999–3005. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.; Casey, M.; Oliva, C.; Newstat, B.; Imwalle, B.; Geyer, C.E. Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: Final survival analysis of a phase III randomized trial. Oncologist 2010, 15, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.; Casey, M.; Press, M.; Lindquist, D.; Pienkowski, T.; Romieu, C.G.; Chan, S.; Jagiello-Gruszfeld, A.; Kaufman, B.; Crown, J.; et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: Updated efficacy and biomarker analyses. Breast Cancer Res. Treat. 2008, 112, 533–543. [Google Scholar] [CrossRef]
- Labonte, M.J.; Wilson, P.M.; Yang, D.; Zhang, W.; Ladner, R.D.; Ning, Y.; Gerger, A.; Bohanes, P.O.; Benhaim, L.; El-Khoueiry, R.; et al. The Cyclin D1 (CCND1) A870G polymorphism predicts clinical outcome to lapatinib and capecitabine in HER2-positive metastatic breast cancer. Ann. Oncol. 2012, 23, 1455–1464. [Google Scholar] [CrossRef]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G.; et al. Neratinib, an Irreversible ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Advanced ErbB2-Positive Breast Cancer. J. Clin. Oncol. 2010, 28, 1301–1307. [Google Scholar] [CrossRef]
- Martin, M.; Bonneterre, J.; Geyer, C.E.; Ito, Y.; Ro, J.; Lang, I.; Kim, S.-B.; Germa, C.; Vermette, J.; Wang, K.; et al. A phase two randomised trial of neratinib monotherapy versus lapatinib plus capecitabine combination therapy in patients with HER2+ advanced breast cancer. Eur. J. Cancer 2013, 49, 3763–3772. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gelmon, K.A.; Verma, S.; Wardley, A.; Conte, P.; Miles, D.; Bianchi, G.; Cortes, J.; McNally, V.A.; Ross, G.A.; et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J. Clin. Oncol. 2010, 28, 1138–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J.; Cortés, J.; Kim, S.-B.; Im, S.-A.; Hegg, R.; Im, Y.-H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II Study of the Antibody Drug Conjugate Trastuzumab-DM1 for the Treatment of Human Epidermal Growth Factor Receptor 2 (HER2) –Positive Breast Cancer After Prior HER2-Directed Therapy. J. Clin. Oncol. 2010, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2019, 382, 597–609. [Google Scholar] [CrossRef]
- Ma, F.; Li, Q.; Chen, S.; Zhu, W.; Fan, Y.; Wang, J.; Luo, Y.; Xing, P.; Lan, B.; Li, M.; et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients with Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2017, 35, 3105–3112. [Google Scholar] [CrossRef]
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or Lapatinib Combined With Capecitabine in HER2–Positive Metastatic Breast Cancer With Prior Taxanes, Anthracyclines, and/or Trastuzumab: A Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 2610–2619. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Sun, T.; Li, W.; Teng, Y.e.; Hu, X.; Bondarenko, I.; Adamchuk, H.; Zhang, L.; Trukhin, D.; et al. Efficacy, Safety, and Immunogenicity of HLX02 Compared with Reference Trastuzumab in Patients with Recurrent or Metastatic HER2-Positive Breast Cancer: A Randomized Phase III Equivalence Trial. BioDrugs 2021. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The Effects of the Novel, Reversible Epidermal Growth Factor Receptor/ErbB-2 Tyrosine Kinase Inhibitor, GW2016, on the Growth of Human Normal and Tumor-derived Cell Lines in Vitro and in Vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the Dual Kinase Inhibitor Lapatinib (GW572016) against HER-2-Overexpressing and Trastuzumab-Treated Breast Cancer Cells. Cancer Res. 2006, 66, 1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.-P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’Brien, N.A.; Roxanis, I.; Li, J.-L.; Bridge, E.; et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhan, D.R.; Guerrero-Zotano, A.; Won, H.; González Ericsson, P.; Servetto, A.; Huerta-Rosario, M.; Ye, D.; Lee, K.-m.; Formisano, L.; Guo, Y.; et al. Hyperactivation of TORC1 Drives Resistance to the Pan-HER Tyrosine Kinase Inhibitor Neratinib in HER2-Mutant Cancers. Cancer Cell 2020, 37, 183–199. [Google Scholar] [CrossRef]
- Chan, A.; Delaloge, S.; Holmes, F.A.; Moy, B.; Iwata, H.; Harvey, V.J.; Robert, N.J.; Silovski, T.; Gokmen, E.; von Minckwitz, G.; et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 367–377. [Google Scholar] [CrossRef]
- Tóth, G.; Szöőr, Á.; Simon, L.; Yarden, Y.; Szöllősi, J.; Vereb, G. The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs 2016, 8, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002, 2, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly Enhanced Antitumor Activity of Trastuzumab and Pertuzumab Combination Treatment on HER2-Positive Human Xenograft Tumor Models. Cancer Res. 2009, 69, 9330. [Google Scholar] [CrossRef] [Green Version]
- Bines, J.; Clark, E.; Barton, C.; Restuccia, E.; Procter, M.; Sonnenblick, A.; Fumagalli, D.; Parlier, D.; Arahmani, A.; Baselga, J.; et al. Patient-reported function, health-related quality of life, and symptoms in APHINITY: Pertuzumab plus trastuzumab and chemotherapy in HER2-positive early breast cancer. Br. J. Cancer 2021. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020, 19, 976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, E.; Xu, Y.; Gao, W.; Wang, L.; Xie, M.H.; Qin, P.; Lu, L.; Li, S.; Shen, P.; et al. Demonstrating Analytical Similarity of Trastuzumab Biosimilar HLX02 to Herceptin(®) with a Panel of Sensitive and Orthogonal Methods Including a Novel FcγRIIIa Affinity Chromatography Technology. BioDrugs 2020, 34, 363–379. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Ding, Y.; Yu, Y.; Wang, M.; Zhou, W.; Wang, J.; Zhu, X.; Zhang, H.; Wang, M.; Chai, K.; et al. A Phase 1 randomized study compare the pharmacokinetics, safety and immunogenicity of HLX02 to reference CN- and EU-sourced trastuzumab in healthy subjects. Cancer Chemother. Pharmacol. 2021, 87, 349–359. [Google Scholar] [CrossRef]
- Andrade-Vieira, R.; Colp, P.; Xu, Z.; Marignani, P.A. Loss of lkb1 expression reduces the latency of ErbB2-mediated mammary gland tumourigenesis, promoting changes in metabolic pathways. PLoS ONE 2013, 8, e56567. [Google Scholar] [CrossRef]
- Paul, M.R.; Pan, T.-c.; Pant, D.K.; Shih, N.N.C.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.D.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Investig. 2020, 130, 4252–4265. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.-i.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (mTOR) Partner, Raptor, Binds the mTOR Substrates p70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos, D.S.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Baas, A.F.; Boudeau, J.; Sapkota, G.P.; Smit, L.; Medema, R.; Morrice, N.A.; Alessi, D.R.; Clevers, H.C. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003, 22, 3062–3072. [Google Scholar] [CrossRef] [Green Version]
- Boudeau, J.; Baas, A.F.; Deak, M.; Morrice, N.A.; Kieloch, A.; Schutkowski, M.; Prescott, A.R.; Clevers, H.C.; Alessi, D.R. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003, 22, 5102–5114. [Google Scholar] [CrossRef] [Green Version]
- Marignani, P.A.; Scott, K.D.; Bagnulo, R.; Cannone, D.; Ferrari, E.; Stella, A.; Guanti, G.; Simone, C.; Resta, N. Novel Splice Isoforms of STRADalpha Differentially Affect LKB1 Activity, Complex Assembly and Subcellular Localization. Cancer Biol. Ther. 2007, 6, 1627–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.-P.; Leiper, F.C.; Woods, A.; Carling, D.; Carlson, M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 8839–8843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.-L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Bell, C.M.; Rothbart, S.B.; Moran, R.G. AMP-activated Protein Kinase (AMPK) Control of mTORC1 Is p53- and TSC2-independent in Pemetrexed-treated Carcinoma Cells. J. Biol. Chem. 2015, 290, 27473–27486. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, B.; Makhdoomi, U.; Vishwakarma, R.; Malik, F. Protein kinase B: Emerging mechanisms of isoform-specific regulation of cellular signaling in cancer. Anti-Cancer Drugs 2017, 28, 569–580. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Reynolds, T.H.; Bodine, S.C.; Lawrence, J.C. Control of Ser2448 Phosphorylation in the Mammalian Target of Rapamycin by Insulin and Skeletal Muscle Load. J. Biol. Chem. 2002, 277, 17657–17662. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.S.; Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Sowadski, J.M. Structural Framework for the Protein Kinase Family. Annu. Rev. Cell Biol. 1992, 8, 429–462. [Google Scholar] [CrossRef]
- Garton, A.J.; Campbell, D.G.; Cohen, P.; Yeaman, S.J. Primary structure of the site on bovine hormone-sensitive lipase phosphorylated by cyclic AMP-dependent protein kinase. FEBS Lett. 1988, 229, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.S.; Weakland, L.L.; Farese, R.V.; West, L.A. Luteinizing hormone increases inositol trisphosphate and cytosolic free Ca2+ in isolated bovine luteal cells. J. Biol. Chem. 1987, 262, 8515–8521. [Google Scholar] [CrossRef]
- Kari, S.; Vasko, V.V.; Priya, S.; Kirschner, L.S. PKA Activates AMPK Through LKB1 Signaling in Follicular Thyroid Cancer. Front. Endocrinol. (Lausanne) 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Fu, V.; Hong, A.W.; Yu, F.-X.; Meng, D.; Melick, C.H.; Wang, H.; Lam, W.-L.M.; Yuan, H.-X.; Taylor, S.S.; et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. Elife 2019, 8, e43038. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 Contributes to the Regulation of Energy Balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Hong, S.-P.; Carlson, M. Mammalian TAK1 Activates Snf1 Protein Kinase in Yeast and Phosphorylates AMP-activated Protein Kinase in vitro*. J. Biol. Chem. 2006, 281, 25336–25343. [Google Scholar] [CrossRef] [Green Version]
- Ovens, A.J.; Scott, J.W.; Langendorf, C.G.; Kemp, B.E.; Oakhill, J.S.; Smiles, W.J. Post-Translational Modifications of the Energy Guardian AMP-Activated Protein Kinase. Int. J. Mol. Sci. 2021, 22, 1229. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.; Richardson, C.; Tee, A.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Averous, J.; Fonseca, B.D.; Proud, C.G. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 2008, 27, 1106–1113. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Kumarasamy, V.; Ruiz, A.; Sivinski, J.; Chung, S.; Grant, A.; Vail, P.; Chauhan, S.S.; Jie, T.; Riall, T.S.; et al. Cell cycle plasticity driven by MTOR signaling: Integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 2019, 38, 3355–3370. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, Y.; Inamitsu, T.; Chida, K.; Iemura, S.-I.; Natsume, T.; Maeda, T.; Hakuno, F.; Takahashi, S.-I. Serine Phosphorylation by mTORC1 Promotes IRS-1 Degradation through SCFβ-TRCP E3 Ubiquitin Ligase. iScience 2018, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal Muscle-Specific Ablation of raptor, but Not of rictor, Causes Metabolic Changes and Results in Muscle Dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, L.-A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Gao, M.; Kong, Q.; Hua, H.; Yin, Y.; Wang, J.; Luo, T.; Jiang, Y. AMPK-mediated up-regulation of mTORC2 and MCL-1 compromises the anti-cancer effects of aspirin. Oncotarget 2016, 7, 16349–16361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, C.; Woo, J.-T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic Disruption of the rictor Gene in Mice Reveals that mTOR Complex 2 Is Essential for Fetal Growth and Viability. Dev. Cell 2006, 11, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.-L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and up-regulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Xu, M.; Liu, P.; Zhang, S.; Shang, R.; Qiao, Y.; Che, L.; Ribback, S.; Cigliano, A.; Evert, K.; et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019, 70, 1600–1613. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.J.; Albers, M.W.; Bum Shin, T.; ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-Rapamycin Complex Interacting with Binding Domain of Human FRAP. Science 1996, 273, 239. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, W.; Serkova, N.; Hausen, B.; Morris, R.E.; Benet, L.Z.; Christians, U. Comparison of the in vitro metabolism of the macrolide immunosuppressants sirolimus and RAD. Transplant. Proc. 2001, 33, 514–515. [Google Scholar] [CrossRef]
- Augustine, J.J.; Hricik, D.E. Experience with everolimus. Transplant. Proc. 2004, 36, S500–S503. [Google Scholar] [CrossRef]
- Dudkin, L.; Dilling, M.B.; Cheshire, P.J.; Harwood, F.C.; Hollingshead, M.; Arbuck, S.G.; Travis, R.; Sausville, E.A.; Houghton, P.J. Biochemical Correlates of mTOR Inhibition by the Rapamycin Ester CCI-779 and Tumor Growth Inhibition. Clin. Cancer Res. 2001, 7, 1758. [Google Scholar] [PubMed]
- Crowe, A.; Bruelisauer, A.; Duerr, L.; Guntz, P.; Lemaire, M. Absorption and Intestinal Metabolism of SDZ-RAD and Rapamycin in Rats. Drug Metab. Dispos. 1999, 27, 627. [Google Scholar] [PubMed]
- Corradetti, M.N.; Guan, K.L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, M.M.; Mita, A.C.; Chu, Q.S.; Rowinsky, E.K.; Fetterly, G.J.; Goldston, M.; Patnaik, A.; Mathews, L.; Ricart, A.D.; Mays, T.; et al. Phase I Trial of the Novel Mammalian Target of Rapamycin Inhibitor Deforolimus (AP23573; MK-8669) Administered Intravenously Daily for 5 Days Every 2 Weeks to Patients with Advanced Malignancies. J. Clin. Oncol. 2008, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2010, 70, 288. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Vieira, R.; Goguen, D.; Bentley, H.A.; Bowen, C.V.; Marignani, P.A. Pre-clinical study of drug combinations that reduce breast cancer burden due to aberrant mTOR and metabolism promoted by LKB1 loss. Oncotarget 2014, 5, 12738–12752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, S.M.; Curwen, J.; Bihani, T.; Cruz, C.M.; Yates, J.W.T.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol. Cancer Ther. 2015, 14, 2508. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Thoreen, C.; Wang, J.; Sabatini, D.; Gray, N.S. mTOR Mediated Anti-Cancer Drug Discovery. Drug Discov. Today Ther. Strateg. 2009, 6, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant Plus Vistusertib vs Fulvestrant Plus Everolimus vs Fulvestrant Alone for Women with Hormone Receptor-Positive Metastatic Breast Cancer: The MANTA Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1556–1564. [Google Scholar] [CrossRef]
- Bendell, J.C.; Kelley, R.K.; Shih, K.C.; Grabowsky, J.A.; Bergsland, E.; Jones, S.; Martin, T.; Infante, J.R.; Mischel, P.S.; Matsutani, T.; et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 2015, 121, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Wolin, E.; Mita, A.; Mahipal, A.; Meyer, T.; Bendell, J.; Nemunaitis, J.; Munster, P.N.; Paz-Ares, L.; Filvaroff, E.H.; Li, S.; et al. A phase 2 study of an oral mTORC1/mTORC2 kinase inhibitor (CC-223) for non-pancreatic neuroendocrine tumors with or without carcinoid symptoms. PLoS ONE 2019, 14, e0221994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.-P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef]
- Carlo, M.I.; Molina, A.M.; Lakhman, Y.; Patil, S.; Woo, K.; DeLuca, J.; Lee, C.-H.; Hsieh, J.J.; Feldman, D.R.; Motzer, R.J.; et al. A Phase Ib Study of BEZ235, a Dual Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in Patients with Advanced Renal Cell Carcinoma. Oncologist 2016, 21, 787–788. [Google Scholar] [CrossRef] [Green Version]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic Characterization of a Potent Inhibitor of Class I Phosphatidylinositide 3-Kinases. Cancer Res. 2007, 67, 5840. [Google Scholar] [CrossRef] [Green Version]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Koch, A.; Evers, B.M. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/Akt/mTOR pathways. Anticancer Res. 2010, 30, 4951–4958. [Google Scholar]
- Andre, F.; Campone, M.; O’Regan, R.; Manlius, C.; Massacesi, C.; Tarek, S.; Mukhopadhyay, P.; Soria, J.-C.; Naughton, M.; Hurvitz, S.A. Phase I Study of Everolimus Plus Weekly Paclitaxel and Trastuzumab in Patients with Metastatic Breast Cancer Pretreated with Trastuzumab. J. Clin. Oncol. 2010, 28, 5110–5115. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Andre, F.; Jiang, Z.; Shao, Z.; Mano, M.S.; Neciosup, S.P.; Tseng, L.-M.; Zhang, Q.; Shen, K.; Liu, D.; et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): A phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015, 16, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Toi, M.; Shao, Z.; Hurvitz, S.; Tseng, L.-M.; Zhang, Q.; Shen, K.; Liu, D.; Feng, J.; Xu, B.; Wang, X.; et al. Efficacy and safety of everolimus in combination with trastuzumab and paclitaxel in Asian patients with HER2+ advanced breast cancer in BOLERO-1. Breast Cancer Res. 2017, 19, 47. [Google Scholar] [CrossRef] [Green Version]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Seiler, M.; Ray-Coquard, I.; Melichar, B.; Yardley, D.A.; Wang, R.X.; Dodion, P.F.; Lee, M.A. Oral Ridaforolimus Plus Trastuzumab for Patients with HER2+ Trastuzumab-Refractory Metastatic Breast Cancer. Clin. Breast Cancer 2015, 15, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Van Swearingen, A.E.D.; Siegel, M.B.; Deal, A.M.; Sambade, M.J.; Hoyle, A.; Hayes, D.N.; Jo, H.; Little, P.; Dees, E.C.; Muss, H.; et al. LCCC 1025: A phase II study of everolimus, trastuzumab, and vinorelbine to treat progressive HER2-positive breast cancer brain metastases. Breast Cancer Res. Treat. 2018, 171, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Karsy, M.; Albert, L.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor Activity of Rapamycin in a Phase I Trial for Patients with Recurrent PTEN-Deficient Glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, N.A.; McDonald, K.; Tong, L.; von Euw, E.; Kalous, O.; Conklin, D.; Hurvitz, S.A.; di Tomaso, E.; Schnell, C.; Linnartz, R.; et al. Targeting PI3K/mTOR Overcomes Resistance to HER2-Targeted Therapy Independent of Feedback Activation of Akt. Clin. Cancer Res. 2014, 20, 3507–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Pérez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharmacol. 2018, 82, 285–298. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Jerusalem, G.; Fasolo, A.; Dieras, V.; Cardoso, F.; Bergh, J.; Vittori, L.; Zhang, Y.; Massacesi, C.; Sahmoud, T.; Gianni, L. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res. Treat. 2011, 125, 447–455. [Google Scholar] [CrossRef]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Hardy, W.R.; Zuo, D.M.; Lam, S.H.; Sanguin-Gendreau, V.; Cardiff, R.D.; Pawson, T.; Muller, W.J. ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J. 2008, 27, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.C.; Chang, Y.-C.; Lu, Y.-S.; Chung, K.-P.; Huang, C.-S.; Lu, T.-P.; Kuo, W.-H.; Wang, M.-Y.; Kuo, K.-T.; Wu, P.-F.; et al. Clinical Relevance of Liver Kinase B1(LKB1) Protein and Gene Expression in Breast Cancer. Sci. Rep. 2016, 6, 21374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entner, N.; Doudoroff, M. Glucose and Gluconic Acid Oxidation of Pseudomonas Saccharophila. J. Biol. Chem. 1952, 196, 853–862. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Bonuccelli, G.; Ozsvári, B.; Peiris-Pagès, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500. [Google Scholar] [CrossRef] [PubMed]
- Morais, R.; Zinkewich-Péotti, K.; Parent, M.; Wang, H.; Babai, F.; Zollinger, M. Tumor-forming Ability in Athymic Nude Mice of Human Cell Lines Devoid of Mitochondrial DNA. Cancer Res. 1994, 54, 3889. [Google Scholar] [PubMed]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.D.; Hagemann, T.; Mather, S.J.; Avril, N. Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death Dis. 2014, 5, e1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sánchez-Martín, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Up-regulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- Cappelletti, V.; Iorio, E.; Miodini, P.; Silvestri, M.; Dugo, M.; Daidone, M.G. Metabolic Footprints and Molecular Subtypes in Breast Cancer. Dis. Markers 2017, 2017, 7687851. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yang, Y.; Qian, K.; Li, L.; Zhang, C.; Fu, X.; Zhang, X.; Chen, H.; Liu, Q.; Cao, S.; et al. A novel tumor suppressor ZBTB1 regulates tamoxifen resistance and aerobic glycolysis through suppressing HER2 expression in breast cancer. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Gale, M.; Li, Y.; Cao, J.; Liu, Z.Z.; Holmbeck, M.A.; Zhang, M.; Lang, S.M.; Wu, L.; Do Carmo, M.; Gupta, S.; et al. Acquired Resistance to HER2-Targeted Therapies Creates Vulnerability to ATP Synthase Inhibition. Cancer Res. 2020, 80, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Hamilton, A.; Geng, S.; Hong, T.; Kalkum, M.; Momand, J.; Kane, S.E.; Huss, J.M. t-Darpp Activates IGF-1R Signaling to Regulate Glucose Metabolism in Trastuzumab-Resistant Breast Cancer Cells. Clin. Cancer Res. 2018, 24, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892. [Google Scholar] [CrossRef] [Green Version]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef] [Green Version]
- Dutchak, P.A.; Estill-Terpack, S.J.; Plec, A.A.; Zhao, X.; Yang, C.; Chen, J.; Ko, B.; Deberardinis, R.J.; Yu, Y.; Tu, B.P. Loss of a Negative Regulator of mTORC1 Induces Aerobic Glycolysis and Altered Fiber Composition in Skeletal Muscle. Cell Rep. 2018, 23, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Decoux-Poullot, A.G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.C.; Krishan, A.; Lampidis, T.J. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-d-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother. Pharmacol. 2004, 53, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Liu, X.; Schafer-Hales, K.; Marcus, A.I.; Khuri, F.R.; Sun, S.-Y.; Zhou, W. 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition. Mol. Cancer Ther. 2008, 7, 809–817. [Google Scholar] [CrossRef] [Green Version]
- Tsurutani, J.; Fukuoka, J.; Tsurutani, H.; Shih, J.H.; Hewitt, S.M.; Travis, W.D.; Jen, J.; Dennis, P.A. Evaluation of Two Phosphorylation Sites Improves the Prognostic Significance of Akt Activation in Non–Small-Cell Lung Cancer Tumors. J. Clin. Oncol. 2006, 24, 306–314. [Google Scholar] [CrossRef]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef]
- DeSalvo, J.; Kuznetsov, J.N.; Du, J.; Leclerc, G.M.; Leclerc, G.J.; Lampidis, T.J.; Barredo, J.C. Inhibition of Akt Potentiates 2-DG–Induced Apoptosis via Down-regulation of UPR in Acute Lymphoblastic Leukemia. Mol. Cancer Res. 2012, 10, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.; Anderson, K.; Franzone, J.; Kemp, B.; Means, A.; Witters, L. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [Green Version]
- Konagaya, Y.; Terai, K.; Hirao, Y.; Takakura, K.; Imajo, M.; Kamioka, Y.; Sasaoka, N.; Kakizuka, A.; Sumiyama, K.; Asano, T.; et al. A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Rep. 2017, 21, 2628–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Tokumitsu, H.; Inuzuka, H.; Murata-Hori, M.; Hosoya, H.; Kobayashi, R. Identification and characterization of novel components of a Ca2+/calmodulin-dependent protein kinase cascade in HeLa cells. FEBS Lett. 2003, 550, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Vijayaraghavan, R.; Kumar, D.; Dube, S.N.; Singh, R.; Pandey, K.S.; Bag, B.C.; Kaushik, M.P.; Sekhar, K.; Dwarakanath, B.S.; Ravindranath, T. Acute toxicity and cardio-respiratory effects of 2-deoxy-D-glucose: A promising radio sensitiser. Biomed. Environ. Sci. 2006, 19, 96–103. [Google Scholar]
- Terse, P.S.; Joshi, P.S.; Bordelon, N.R.; Brys, A.M.; Patton, K.M.; Arndt, T.P.; Sutula, T.P. 2-Deoxy-d-Glucose (2-DG)-Induced Cardiac Toxicity in Rat: NT-proBNP and BNP as Potential Early Cardiac Safety Biomarkers. Int. J. Toxicol. 2016, 35, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-Deoxy-D-glucose Increases the Efficacy of Adriamycin and Paclitaxel in Human Osteosarcoma and Non-Small Cell Lung Cancers In Vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, X.; Fu, H.; Zhou, W.; Zhong, D. 2-Deoxyglucose Suppresses ERK Phosphorylation in LKB1 and Ras Wild-Type Non-Small Cell Lung Cancer Cells. PLoS ONE 2016, 11, e0168793. [Google Scholar] [CrossRef]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-D-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [CrossRef]
- Ma, S.; Jia, R.; Li, D.; Shen, B. Targeting Cellular Metabolism Chemosensitizes the Doxorubicin-Resistant Human Breast Adenocarcinoma Cells. BioMed. Res. Int. 2015, 2015, 453986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guldner, I.H.; Golomb, S.M.; Sun, L.; Harris, J.A.; Lu, X.; Zhang, S. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat. Commun. 2019, 10, 3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Diéras, V.; Im, S.-A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, M.; Cristaudo, A.; Morrone, A.; Messina, C.; Bennardo, L.; Nisticò, S.P.; Mariano, M.; Cameli, N. Emerging Skin Toxicities in Patients with Breast Cancer Treated with New Cyclin-Dependent Kinase 4/6 Inhibitors: A Systematic Review. Drug Saf. 2021. [Google Scholar] [CrossRef]


| Treatment | Trial Name | Patients | ORR | Median PFS (Months) | OS (Months) | REF | |
|---|---|---|---|---|---|---|---|
| Trastuzumab (T) | Monotherapy | Phase II | 48 | 11.60% | 5.1 | n/a | [32] |
| T, chemotherapy | n/a | 90 | 49.50% | 10 | 16 (from first TZB treatment) | [34] | |
| T, cisplatin, or carboplatin | UCLA-ORN/BCIRG Phase III | 62 | 58% | 12.7 | OS not reached | [33] | |
| Lapatinib (L) | Monotherapy | EGF20009 Phase II | 136 | 24–31% | 43% of patients at 6 months | OS not reached | [41] |
| capecitabine (cap) | EGF100151 (NCT00078572) Phase III | 399 | 13.90% | 4.4 | 15.3 | [42,43,44,45] | |
| L, cap | 23.7% | 8.4 | 15.6 | ||||
| Neratinib (N) | Monotherapy (prior T therapy) | NCT00300781 Phase II | 66 | 24% | 22.3 | n/a | [46] |
| Monotherapy (no prior therapy) | 70 | 56% | 39.6 | n/a | |||
| Monotherapy | NCT00777101 Phase II | 117 | 29% | 4.5 | 19.7 | [47] | |
| N, cap | NALA (NCT01808573) Phase III | 621 | 32.80% | 5.6 | 24 | [48] | |
| Pertuzumab (PRT) | PRT, T | NCT03001899 Phase II | 66 | 34% | 5.5 | n/a | [49] |
| PRT, T, docetaxel | CLEOPATRA (NCT00567190) Phase III | 808 | 80.20% | 18.7 | 56.5 | [50,51] | |
| PZB, T, chemotherapy | APHINITY (NCT01358877) Phase III | 4805 | n/a | 94.1% of patients with IDFS at 3 years | n/a | [52] | |
| T-DM1 | Monotherapy | Phase II | 112 | 25.90% | 4.6 | n/a | [53] |
| Monotherapy | EMILA (NCT00829166) Phase III | 991 | 43.60% | 9.5 | 30.9 | [54] | |
| T-deruxtecan (DS-8201; DS) | Monotherapy (prior therapies) | DESTINY-Breast01 (NCT03248492) Phase II | 184 | 60.90% | 16.4 | OS not reached | [55] |
| Tucatinib (TN) | TN, T, cap | HER2CLIMB (NCT02614794) Phase II | 612 | 40.60% | 7.8 | 44.9% at 2 years | [56] |
| Pyrotinib (P) | P, cap (prior therapies) | NCT01937689 Phase I | 38 | 50.00% | 35.4 | n/a | [57] |
| P, cap | NCT02422199 Phase II | 128 | 78.50% | 18.1 | n/a | [58] | |
| HLX02 | HLX02, docetaxel | NCT03084237 Phase III | 649 | 71.30% | 11.7 | n/a | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holloway, R.W.; Marignani, P.A. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers 2021, 13, 2922. https://doi.org/10.3390/cancers13122922
Holloway RW, Marignani PA. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers. 2021; 13(12):2922. https://doi.org/10.3390/cancers13122922
Chicago/Turabian StyleHolloway, Ryan W., and Paola A. Marignani. 2021. "Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer" Cancers 13, no. 12: 2922. https://doi.org/10.3390/cancers13122922
APA StyleHolloway, R. W., & Marignani, P. A. (2021). Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers, 13(12), 2922. https://doi.org/10.3390/cancers13122922