
Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy?


Simple Summary
Abstract
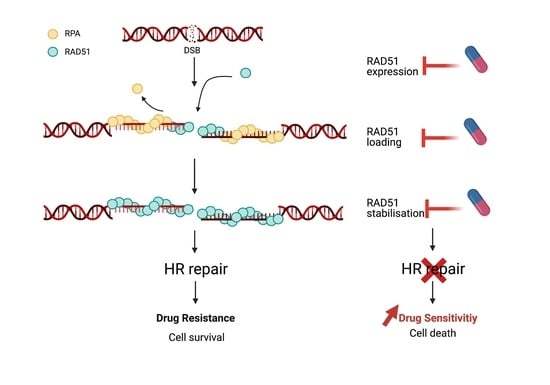
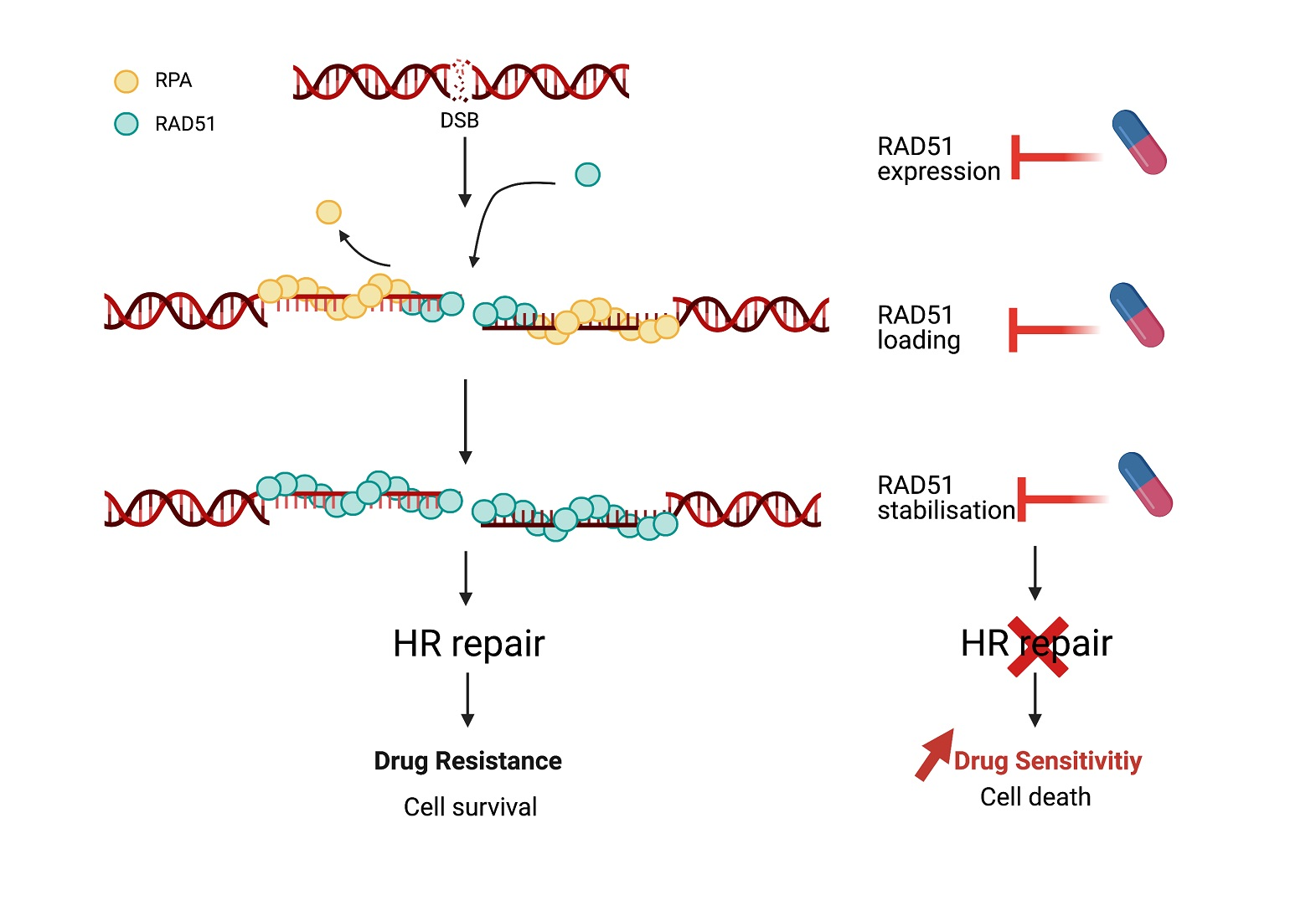
1. Introduction
2. Regulation of RAD51 Expression
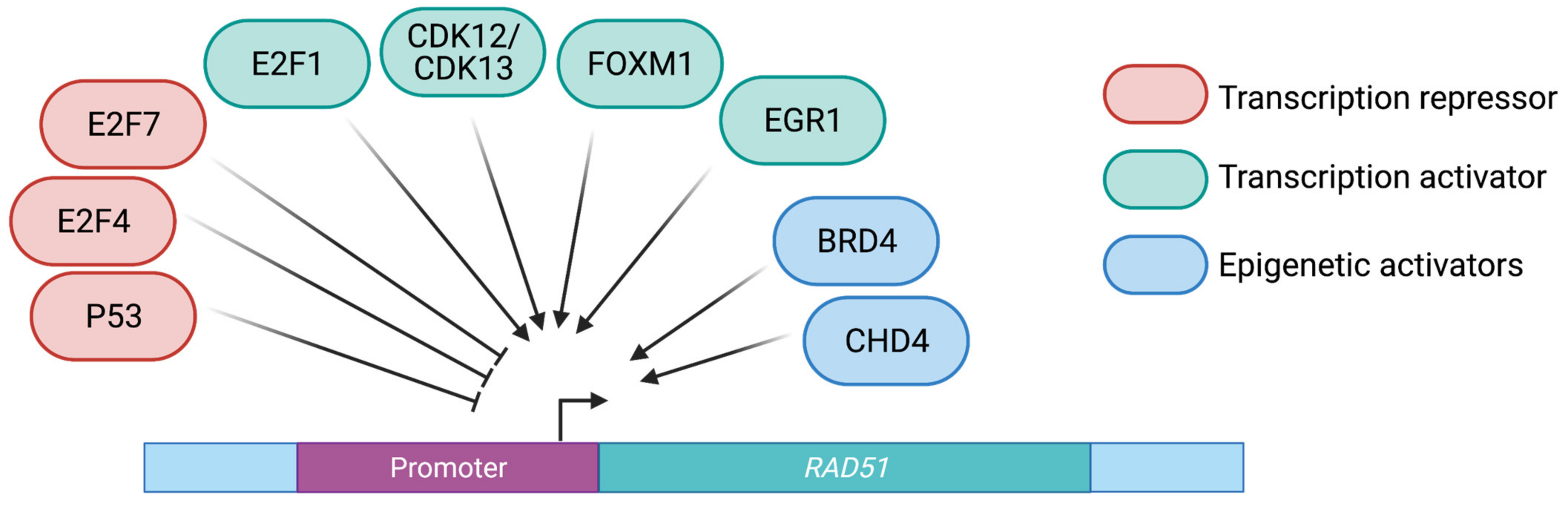
2.1. Transcriptional Control of RAD51
2.1.1. Cyclin Dependent Kinases
2.1.2. Member of the E2F Transcription Factor Family
2.1.3. Other Transcription Factors
2.2. Chromatin-Mediated Regulation of RAD51 Gene Expression
3. Post-Translational Regulation of RAD51
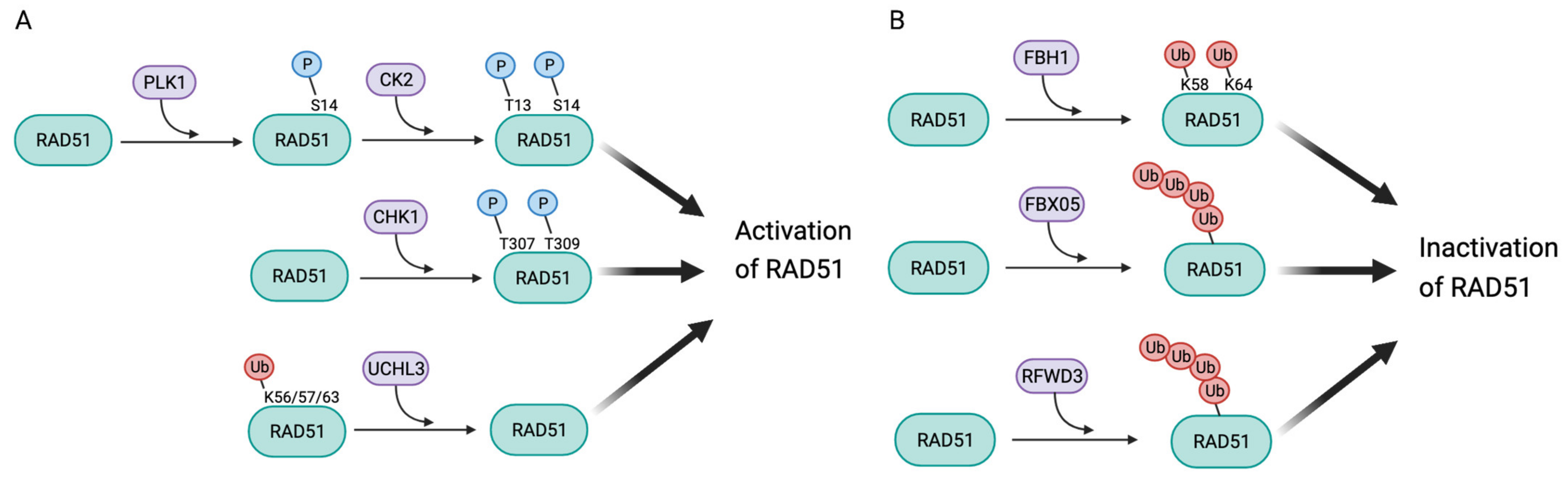
3.1. Post-Translational Modifications Leading to Activation of RAD51
3.2. Post-Translational Modifications Leading to RAD51 Inactivation
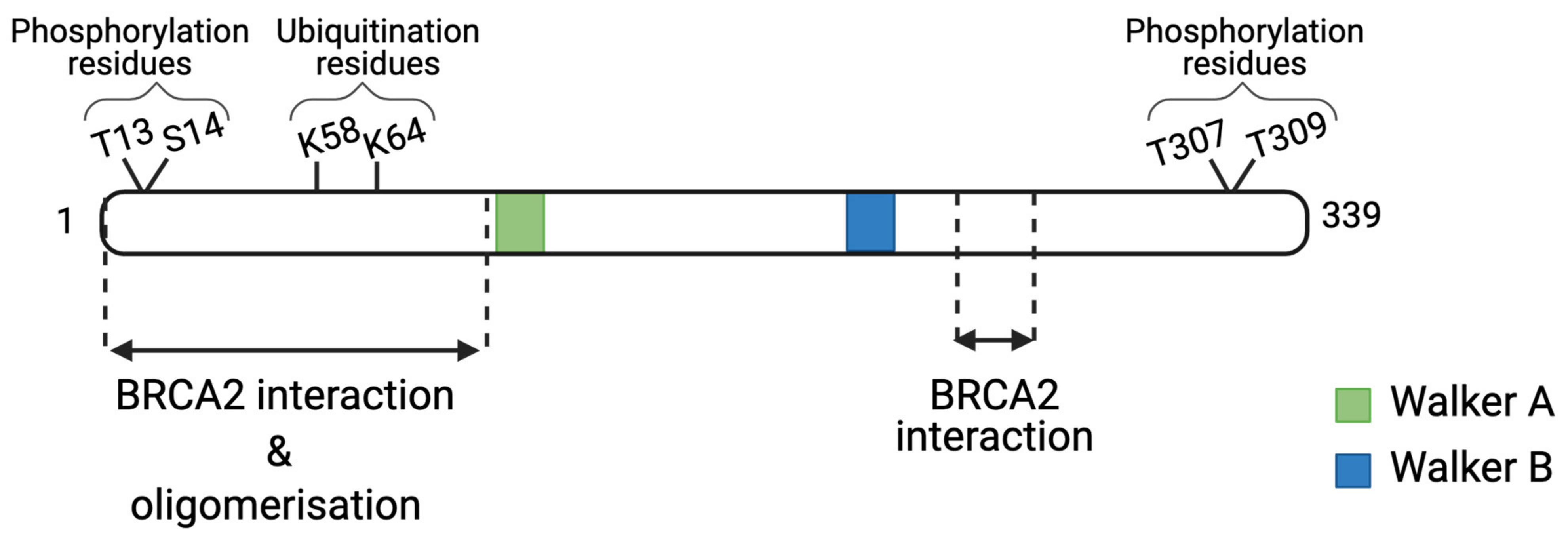
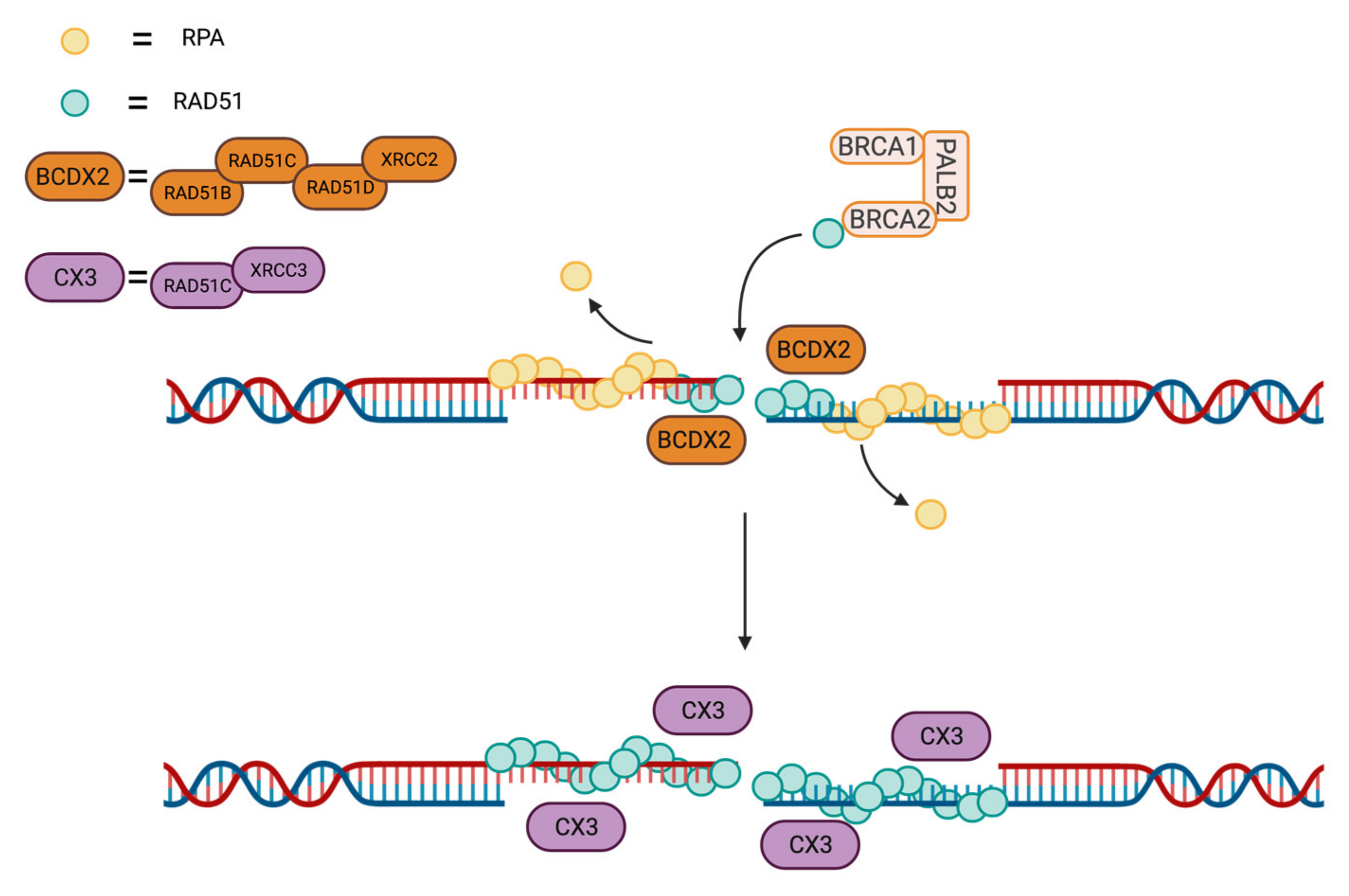
3.3. RAD51 Loading and Stability at DSB Site by Protein–Protein Interactions
4. Pharmacological Inhibition of RAD51
5. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia-Telangiectasia Mutated |
| ATR | Ataxia Telangiectasia and Rad3 Related |
| BD | Bromodomain |
| BET | Bromodomains and Extraterminal |
| BRCA1 | Breast Cancer 1 |
| BRCT | BRCA1 Carboxy-Terminus |
| CCNK | Cyclin K |
| CDK | Cyclin-Dependent Kinase |
| ChIP | Chromatin Immunoprecipitation |
| CHD4 | Chromodomain Helicase DNA-binding Protein 4 |
| CK2 | Casein Kinase 2 |
| CML | Chronic Myelogenous Leukemia |
| CTD | C-Terminal Domain |
| CtIP | CtBP-Interacting Protein |
| DNA | Deoxyribonucleic acid (necessary?) |
| DNA-PKcs | DNA-dependent Protein Kinase Catalytic Subunit |
| DSB | Double-Strand Break |
| ET | Extraterminal Domain |
| GBM | Glioblastoma Multiforme |
| H2AX | Histone 2AX |
| HR | Homologous Recombination |
| HRR | Homologous Recombination Repair |
| H3K9Ac | Histone H3 at lysine 9 |
| IR | Ionizing Radiation |
| NHEJ | Non-Homologous End Joining |
| PALB2 | Partner and Localizer of BRCA2 |
| PARPi | Poly (ADP-ribose) Polymerase Inhibitors |
| PLK1 | Polo-like kinase 1 |
| RB | Retinoblastoma |
| RPA | Replication Protein A |
| SE | Super-Enhancers |
| ssDNA | Single-Strand DNA |
| TCGA | The Cancer Genome Atlas |
| UV | Ultra-violet |
| 53BP1 | TP53-binding protein |
References
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.-Y.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. MCB 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Póti, Á.; Gyergyák, H.; Németh, E.; Rusz, O.; Tóth, S.; Kovácsházi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Löbrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.-C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell. Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef]
- Amunugama, R.; Fishel, R. Homologous Recombination in Eukaryotes. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2012; Volume 110, pp. 155–206. ISBN 978-0-12-387665-2. [Google Scholar]
- Jiang, S.; Lin, T.; Xie, Q.; Wang, L. Network analysis of RAD51 proteins in metazoa and the evolutionary relationships with their archaeal homologs. Front. Genet. 2018, 9, 383. [Google Scholar] [CrossRef]
- Suzuki, Y.; Chew, M.L.; Suzuki, Y. Role of host-encoded proteins in restriction of retroviral integration. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein Dynamics of human RPA and RAD51 on SsDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Stark, J.M.; Ommundsen, M.; Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004, 23, 546–553. [Google Scholar] [CrossRef]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Krüger, S.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Sarwar, R.; Sheikh, A.K.; Mahjabeen, I.; Bashir, K.; Saeed, S.; Kayani, M.A. Upregulation of RAD51 expression is associated with progression of thyroid carcinoma. Exp. Mol. Pathol. 2017, 102, 446–454. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, N.; Yao, W.; Li, S.; Ren, Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019, 19, 356. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.-Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T.; et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.-B.; Wu, Y.-L.; Yang, X.-N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef]
- Siaud, N.; Barbera, M.A.; Egashira, A.; Lam, I.; Christ, N.; Schlacher, K.; Xia, B.; Jasin, M. Plasticity of BRCA2 function in homologous recombination: Genetic interactions of the PALB2 and DNA binding domains. PLoS Genet. 2011, 7, e1002409. [Google Scholar] [CrossRef]
- Zhao, Q.; Guan, J.; Zhang, Z.; Lv, J.; Wang, Y.; Liu, L.; Zhou, Q.; Mao, W. Inhibition of Rad51 sensitizes breast cancer cells with wild-type PTEN to olaparib. Biomed. Pharmacother. 2017, 94, 165–168. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Kuo, Y.-H.; Chiu, Y.-F.; Su, Y.-C.; Lin, Y.-W. Down-regulation of Rad51 expression overcomes drug resistance to gemcitabine in human non-small-cell lung cancer cells. J. Pharmacol. Exp. Ther. 2010, 335, 830–840. [Google Scholar] [CrossRef]
- Banerjee, S.; Kaye, S.B.; Ashworth, A. Making the best of PARP inhibitors in ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 508–519. [Google Scholar] [CrossRef]
- Meijer, T.G.; Verkaik, N.S.; van Deurzen, C.H.M.; Dubbink, H.-J.; den Toom, T.D.; Sleddens, H.F.B.M.; De Hoop, E.O.; Dinjens, W.N.M.; Kanaar, R.; van Gent, D.C.; et al. Direct ex vivo observation of homologous recombination defect reversal after DNA-damaging chemotherapy in patients with metastatic breast cancer. JCO Precis. Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Harlen, K.M.; Churchman, L.S. The code and beyond: Transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017, 18, 263–273. [Google Scholar] [CrossRef]
- Eick, D.; Geyer, M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev. 2013, 113, 8456–8490. [Google Scholar] [CrossRef]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 Is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Greenleaf, A.L. Phosphorylation of RNAPII: To P-TEFb or not to P-TEFb? Transcription 2011, 2, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in c-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef] [PubMed]
- Reimers, M.A.; Yip, S.M.; Zhang, L.; Cieslik, M.; Dhawan, M.; Montgomery, B.; Wyatt, A.W.; Chi, K.N.; Small, E.J.; Chinnaiyan, A.M.; et al. Clinical outcomes in cyclin-dependent kinase 12 mutant advanced prostate cancer. Eur. Urol. 2020, 77, 333–341. [Google Scholar] [CrossRef]
- Fusco, N.; Geyer, F.C.; De Filippo, M.R.; Martelotto, L.G.; Ng, C.K.Y.; Piscuoglio, S.; Guerini-Rocco, E.; Schultheis, A.M.; Fuhrmann, L.; Wang, L.; et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod. Pathol. 2016, 29, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Boeva, V.; Battistella, A.; Goundiam, O.; Smith, N.K.; Mueller, C.R.; Raynal, V.; Mariani, O.; Sastre-Garau, X.; et al. Ovarian cancers harboring inactivating mutations in CDK12 display a distinct genomic instability pattern characterized by large tandem duplications. Cancer Res. 2016, 76, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Naidoo, K.; Wai, P.T.; Maguire, S.L.; Daley, F.; Haider, S.; Kriplani, D.; Campbell, J.; Mirza, H.; Grigoriadis, A.; Tutt, A.; et al. Evaluation of CDK12 protein expression as a potential novel biomarker for DNA damage response-targeted therapies in breast cancer. Mol. Cancer Ther. 2018, 17, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Pospichalova, V.; Bösken, C.A.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015, 43, 2575–2589. [Google Scholar] [CrossRef]
- Lui, G.Y.L.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.G.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef]
- Baldwin, P.; Orriols, A.; Sridhar, S. Abstract 3642: Combination Nanotherapy Using the PARP Inhibitor Talazoparib and Cyclin Dependent Kinase Inhibitor Dinaciclib. Cancer Res. 2019, 79, 3642. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Do, K.T.; Tolaney, S.M.; Hilton, J.F.; Cleary, J.M.; Wolanski, A.; Beardslee, B.; Hassinger, F.; Bhushan, K.; Cai, D.; et al. Abstract CT047: Phase 1 Dose-Escalation Study of the CDK Inhibitor Dinaciclib in Combination with the PARP Inhibitor Veliparib in Patients with Advanced Solid Tumors. Cancer Res. 2017, 77, CT047. [Google Scholar] [CrossRef]
- Smith, E.J.; Leone, G.; Nevins, J.R. Distinct mechanisms control the accumulation of the Rb-Related P107 and P130 proteins during cell growth. Cell Growth Differ. 1998, 9, 297–303. [Google Scholar]
- Ikeda, M.A.; Jakoi, L.; Nevins, J.R. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 3215–3220. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-C.; Lin, F.-T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar]
- Stevens, C.; Smith, L.; La Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 2003, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Kotsinas, A.; Bouda, M.; Kletsas, D.; Asimacopoulos, P.J.; Agnantis, N.; Kittas, C.; Papavassiliou, A.G. Transcription factor E2F-1 acts as a growth-promoting factor and is associated with adverse prognosis in non-small cell lung carcinomas. J. Pathol. 2002, 198, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yasui, W.; Yokozaki, H.; Naka, K.; Ishikawa, T.; Tahara, E. Expression of the E2F Family in human gastrointestinal carcinomas. Int. J. Cancer 1999, 81, 535–538. [Google Scholar] [CrossRef]
- Choi, E.-H.; Kim, K.P. E2F1 Facilitates DNA break repair by localizing to break sites and enhancing the expression of homologous recombination factors. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Yang, S.-Z.; Lin, F.-T.; Lin, W.-C. MCPH1/BRIT1 Cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis. EMBO Rep. 2008, 9, 907–915. [Google Scholar] [CrossRef]
- Bindra, R.S.; Glazer, P.M. Repression of RAD51 gene expression by E2F4/P130 complexes in hypoxia. Oncogene 2007, 26, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Mitxelena, J.; Apraiz, A.; Vallejo-Rodríguez, J.; García-Santisteban, I.; Fullaondo, A.; Alvarez-Fernández, M.; Malumbres, M.; Zubiaga, A.M. An E2F7-dependent transcriptional program modulates DNA damage repair and genomic stability. Nucleic Acids Res. 2018, 46, 4546–4559. [Google Scholar] [CrossRef]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.-G.; Moldovan, G.-L. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef]
- Fagan, R.; Flint, K.J.; Jones, N. Phosphorylation of E2F-1 modulates its interaction with the retinoblastoma gene product and the adenoviral E4 19 KDa protein. Cell 1994, 78, 799–811. [Google Scholar] [CrossRef]
- Martínez-Balbás, M.A.; Bauer, U.M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wu, X.; Yang, L.; Xiao, F.; Zhang, H.; Zhou, A.; Huang, Z.; Huang, S. FoxM1 Inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA repair gene Rad51. Clin. Cancer Res. 2012, 18, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Hine, C.M.; Li, H.; Xie, L.; Mao, Z.; Seluanov, A.; Gorbunova, V. Regulation of Rad51 promoter. Cell Cycle 2014, 13, 2038–2045. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. P53 Modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Chiang, C.-M. The Double Bromodomain-Containing Chromatin Adaptor Brd4 and Transcriptional Regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin–remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Rendtlew Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010, 190, 731–740. [Google Scholar] [CrossRef]
- McKenzie, L.D.; LeClair, J.W.; Miller, K.N.; Strong, A.D.; Chan, H.L.; Oates, E.L.; Ligon, K.L.; Brennan, C.W.; Chheda, M.G. CHD4 Regulates the DNA damage response and RAD51 expression in glioblastoma. Sci. Rep. 2019, 9, 4444. [Google Scholar] [CrossRef]
- Ogiwara, H.; Kohno, T. CBP and P300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes. PLoS ONE 2012, 7, e52810. [Google Scholar] [CrossRef]
- Qi, W.; Chen, H.; Xiao, T.; Wang, R.; Li, T.; Han, L.; Zeng, X. Acetyltransferase P300 collaborates with chromodomain helicase DNA-binding protein 4 (CHD4) to facilitate DNA double-strand break repair. Mutagenesis 2016, 31, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.-Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Montenarh, M. Protein kinase CK2 in DNA damage and repair. Transl. Cancer Res. 2016, 5. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in cancer therapy: From laboratory to clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy–potential clinical relevance. Cell Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef]
- Parsels, L.A.; Morgan, M.A.; Tanska, D.M.; Parsels, J.D.; Palmer, B.D.; Booth, R.J.; Denny, W.A.; Canman, C.E.; Kraker, A.J.; Lawrence, T.S.; et al. Gemcitabine sensitization by Chk1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol. Cancer Ther. 2009, 8, 45–54. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between HBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Viñolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Corral Jaime, J.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef]
- Banerji, U.; Plummer, E.R.; Moreno, V.; Ang, J.E.; Quinton, A.; Drew, Y.; Hernández, T.; Roda, D.; Carter, L.; Navarro, A.; et al. A phase I/II first-in-human trial of oral SRA737 (a Chk1 Inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. JCO 2019, 37, 3095. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Li, L.; Li, Y.; Wu, C.; Yin, Y.; Chen, Y.; Deng, M.; Nowsheen, S.; Yuan, J.; Lou, Z. A phosphorylation–deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Genes Dev. 2016, 30, 2581–2595. [Google Scholar] [CrossRef] [PubMed]
- Osman, F.; Dixon, J.; Barr, A.R.; Whitby, M.C. The f-box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 2005, 25, 8084–8096. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, C.; Morishita, T.; Shinagawa, H.; Hishida, T. Essential and distinct roles of the f-box and helicase domains of Fbh1 in DNA damage repair. BMC Mol. Biol. 2008, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Mistrik, M.; Danielsen, J.R.; Dinant, C.; Falck, J.; Bartek, J.; Lukas, J.; Mailand, N. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J. Cell Biol. 2009, 186, 655–663. [Google Scholar] [CrossRef]
- Lorenz, A.; Osman, F.; Folkyte, V.; Sofueva, S.; Whitby, M.C. Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol. Cell. Biol. 2009, 29, 4742–4756. [Google Scholar] [CrossRef] [PubMed]
- Simandlova, J.; Zagelbaum, J.; Payne, M.J.; Chu, W.K.; Shevelev, I.; Hanada, K.; Chatterjee, S.; Reid, D.A.; Liu, Y.; Janscak, P.; et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 2013, 288, 34168–34180. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The f-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol. Cell 2019, 73, 224–237.e6. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Chen, J. E3 ligase RFWD3 participates in replication checkpoint control. J. Biol. Chem. 2011, 286, 22308–22313. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chu, J.; Yucer, N.; Leng, M.; Wang, S.-Y.; Chen, B.P.C.; Hittelman, W.N.; Wang, Y. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein a (RPA) and facilitates RPA-mediated DNA damage response. J. Biol. Chem. 2011, 286, 22314–22322. [Google Scholar] [CrossRef]
- Inano, S.; Sato, K.; Katsuki, Y.; Kobayashi, W.; Tanaka, H.; Nakajima, K.; Nakada, S.; Miyoshi, H.; Knies, K.; Takaori-Kondo, A.; et al. RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol. Cell 2017, 66, 622–634.e8. [Google Scholar] [CrossRef]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Rep. 2018, 25, 1384. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Larsen, B.D.; Achanta, K.; Sørensen, C.S. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep. 2016, 17, 671–681. [Google Scholar] [CrossRef]
- Buisson, R.; Joshi, N.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.-L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of homologous recombination and the checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Typas, D.; Caron, M.-C.; Wiegant, W.W.; van den Heuvel, D.; Boonen, R.A.; Couturier, A.M.; Mullenders, L.H.; Masson, J.-Y.; van Attikum, H. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 2017, 6, e20922. [Google Scholar] [CrossRef]
- Lin, Z.; Kong, H.; Nei, M.; Ma, H. Origins and evolution of the RecA/RAD51 gene family: Evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 10328–10333. [Google Scholar] [CrossRef]
- Thacker, J. A surfeit of RAD51-like genes? Trends Genet. 1999, 15, 166–168. [Google Scholar] [CrossRef]
- Liu, N.; Lamerdin, J.E.; Tebbs, R.S.; Schild, D.; Tucker, J.D.; Shen, M.R.; Brookman, K.W.; Siciliano, M.J.; Walter, C.A.; Fan, W.; et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol. Cell 1998, 1, 783–793. [Google Scholar] [CrossRef]
- Sung, P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein a to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997, 11, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Fukushima, T.; Morrison, C.; Albala, J.S.; Swagemakers, S.M.A.; Kanaar, R.; Thompson, L.H.; Takeda, S. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell Biol. 2000, 20, 6476–6482. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Tachiiri, S.; Fukushima, T.; Sonoda, E.; Schild, D.; Thompson, L.H.; Takeda, S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell Biol. 2001, 21, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Fuller, L.F.; Painter, R.B. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat. Res. DNA Repair Rep. 1988, 193, 109–121. [Google Scholar] [CrossRef]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef]
- Park, D.J.; Lesueur, F.; Nguyen-Dumont, T.; Pertesi, M.; Odefrey, F.; Hammet, F.; Neuhausen, S.L.; John, E.M.; Andrulis, I.L.; Terry, M.B.; et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am. J. Hum. Genet. 2012, 90, 734–739. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Breast Cancer Susceptibility Collaboration (UK); Eccles, D.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A population-based study of genes previously implicated in breast cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Felicio, P.S.; Grasel, R.S.; Campacci, N.; de Paula, A.E.; Galvão, H.C.R.; Torrezan, G.T.; Sabato, C.S.; Fernandes, G.C.; Souza, C.P.; Michelli, R.D.; et al. Whole-exome sequencing of Non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021, 42, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Llovet, P.; Díez-Gómez, B.; Caloca, M.J.; Pérez-Segura, P.; Fraile-Bethencourt, E.; Colmena, M.; Carvalho, S.; et al. Comprehensive functional characterization and clinical interpretation of 20 splice-site variants of the RAD51C gene. Cancers 2020, 12, 3771. [Google Scholar] [CrossRef]
- Masson, J.Y.; Tarsounas, M.C.; Stasiak, A.Z.; Stasiak, A.; Shah, R.; McIlwraith, M.J.; Benson, F.E.; West, S.C. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001, 15, 3296–3307. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.-Y.; Stasiak, A.Z.; Stasiak, A.; Benson, F.E.; West, S.C. Complex formation by the human RAD51C and XRCC3 recombination repair proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 8440–8446. [Google Scholar] [CrossRef] [PubMed]
- Yonetani, Y.; Hochegger, H.; Sonoda, E.; Shinya, S.; Yoshikawa, H.; Takeda, S.; Yamazoe, M. Differential and collaborative actions of Rad51 paralog proteins in cellular response to DNA damage. Nucleic Acids Res. 2005, 33, 4544–4552. [Google Scholar] [CrossRef]
- Shor, E.; Weinstein, J.; Rothstein, R. A Genetic screen for top3 suppressors in saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: Four genes involved in error-free DNA repair. Genetics 2005, 169, 1275–1289. [Google Scholar] [CrossRef]
- Liu, T.; Wan, L.; Wu, Y.; Chen, J.; Huang, J. HSWS1·SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J. Biol. Chem. 2011, 286, 41758–41766. [Google Scholar] [CrossRef]
- Martín, V.; Chahwan, C.; Gao, H.; Blais, V.; Wohlschlegel, J.; Yates, J.R.; McGowan, C.H.; Russell, P. Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J. 2006, 25, 2564–2574. [Google Scholar] [CrossRef]
- Garcin, E.B.; Gon, S.; Sullivan, M.R.; Brunette, G.J.; Cian, A.D.; Concordet, J.-P.; Giovannangeli, C.; Dirks, W.G.; Eberth, S.; Bernstein, K.A.; et al. Differential requirements for the RAD51 paralogs in genome repair and maintenance in human cells. PLoS Genet. 2019, 15, e1008355. [Google Scholar] [CrossRef]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.G.; Zhang, K.; Cobb, J.A.; Boone, C.; Xiao, W. The Yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol. Microbiol. 2009, 73, 89–102. [Google Scholar] [CrossRef]
- Godin, S.; Wier, A.; Kabbinavar, F.; Bratton-Palmer, D.S.; Ghodke, H.; Van Houten, B.; VanDemark, A.P.; Bernstein, K.A. The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic Acids Res. 2013, 41, 4525–4534. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Zhang, Z.; Herken, B.W.; Westmoreland, J.W.; Lee, A.G.; Mihalevic, M.J.; Yu, Z.; Sobol, R.W.; Resnick, M.A.; Bernstein, K.A. The Shu complex promotes error-free tolerance of alkylation-induced base excision repair products. Nucleic Acids Res. 2016, 44, 8199–8215. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Dejsuphong, D.; Petalcorin, M.I.R.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef]
- Wu, L.; Davies, S.L.; Levitt, N.C.; Hickson, I.D. Potential role for the BLM Helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 2001, 276, 19375–19381. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazin, A.V. A small molecule inhibitor of human RAD51 potentiates breast cancer cell killing by therapeutic agents in mouse xenografts. PLoS ONE 2014, 9, e100993. [Google Scholar] [CrossRef]
- Ward, A.; Dong, L.; Harris, J.M.; Khanna, K.K.; Al-Ejeh, F.; Fairlie, D.P.; Wiegmans, A.P.; Liu, L. Quinazolinone derivatives as inhibitors of homologous recombinase RAD51. Bioorganic Med. Chem. Lett. 2017, 27, 3096–3100. [Google Scholar] [CrossRef]
- Budke, B.; Logan, H.L.; Kalin, J.H.; Zelivianskaia, A.S.; Cameron McGuire, W.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef]
- Budke, B.; Kalin, J.H.; Pawlowski, M.; Zelivianskaia, A.S.; Wu, M.; Kozikowski, A.P.; Connell, P.P. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring michael acceptor reactivity. J. Med. Chem. 2013, 56, 254–263. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, L.; Wu, G.; Konig, H.; Lin, X.; Li, G.; Qiu, X.-L.; Chen, C.-F.; Hu, C.-M.; Goldblatt, E.; et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013, 5, 353–365. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, H.; Guo, X.E.; Qiu, X.-L.; Hu, C.-M.; Chamberlin, A.R.; Lee, W.-H. Synthesis, molecular modeling, and biological evaluation of novel RAD51 inhibitors. Eur. J. Med. Chem. 2015, 96, 196–208. [Google Scholar] [CrossRef]
- Guy, J.L.; Maclay, T.; Day, M.; Burness, M.L.; Mills, K. Abstract P2-05-05: RAD51 Inhibition Using CYT-0851, Shows Anti-Cancer Activity in Cellular Models of Breast Cancer and Acts Synergistically with PARP Inhibitors. Cancer Res. 2020, 80, P2-05-05. [Google Scholar] [CrossRef]
- Saeki, H.; Jogo, T.; Kawazoe, T.; Kamori, T.; Nakaji, Y.; Zaitsu, Y.; Fujiwara, M.; Baba, Y.; Nakamura, T.; Iwata, N.; et al. RAD51 expression as a biomarker to predict efficacy of preoperative therapy and survival for esophageal squamous cell carcinoma: A large-cohort observational study (KSCC1307). Ann. Surg. 2020. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Pataer, A.; Shao, R.; Correa, A.M.; Behrens, C.; Roth, J.A.; Vaporciyan, A.A.; Wistuba, I.I.; Swisher, S.G. Major pathologic response and RAD51 predict survival in lung cancer patients receiving neoadjuvant chemotherapy. Cancer Med. 2018, 7, 2405–2414. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Tanaka, N.; Gleber-Netto, F.O.; Bartels, M.D.; Wang, L.; McGrail, D.J.; Lin, S.-Y.; Frank, S.J.; et al. Combined inhibition of Rad51 and Wee1 enhances cell killing in HNSCC through induction of apoptosis associated with excessive DNA damage and replication stress. Mol. Cancer Ther. 2021, 1535–7163, Online ahead of print. [Google Scholar] [CrossRef]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional genomics identifies new synergistic therapies for retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulator of RAD51 | Function | Level of regulation | Effect on RAD51 | Drug Target |
|---|---|---|---|---|
| CDK12/CDK13 | RNA pol II regulator | mRNA expression | Activator | Yes |
| E2F1 | Transcription Factor | mRNA expression | Activator | No |
| E2F4 | Transcription Factor | mRNA expression | Repressor | No |
| E2F7 | Transcription Factor | mRNA expression | Repressor | No |
| FOXM1 | Transcription Factor | mRNA expression | Activator | No |
| EGR1 | Transcription Factor | mRNA expression | Activator | No |
| P53 | Transcription Factor | mRNA expression | Repressor | No |
| BRD4 | Transcription Activator | mRNA expression | Activator | Yes |
| CHD4 | Nucleosome Remodeler | mRNA expression | Activator | No |
| PLK1 | Kinase | Post-translational | Activator | Yes |
| CK2 | Kinase | Post-translational | Activator | Yes |
| CHK1 | Kinase | Post-translational | Activator | Yes |
| UCHL3 | Deubiquitinatinase | Post-translational | Activator | No |
| FBH1 | Ubiquitin ligase | Post-translational | Repressor | No |
| FBX05 | Ubiquitin ligase | Post-translational | Repressor | No |
| RFWD3 | Ubiquitin ligase | Post-translational | Repressor | No |
| RAD51C | RAD51 Cofactor | Homofilament Loading/stabilisation | Activator | No |
| RAD51B | RAD51 Cofactor | Homofilament Loading | Activator | No |
| RAD51D | RAD51 Cofactor | Homofilament Loading | Activator | No |
| XRCC1 | RAD51 Cofactor | Homofilament stabilisation | Activator | No |
| XRCC2 | RAD51 Cofactor | Homofilament Loading | Activator | No |
| SWSAP1 | RAD51 Cofactor | Homofilament stabilisation | Activator | No |
| PARI | DNA Helicase | Homofilament stabilisation | Repressor | No |
| BLM | DNA Helicase | Homofilament stabilisation | Repressor | No |
| Drug | Target | Trial phase | Trial Status | Results | Trial Number |
|---|---|---|---|---|---|
| CYT-0851 | RAD51 | Phase 1/2 | On going | Not reported | NCT03997968 |
| Dinaciclib | Inhibition of multiple CDKs | Phase 1 | On going | Tolerable in combination with veliparib | NCT01434316 |
| Anti-tumor activity is limited in non-BRCA carriers | |||||
| INCB054329 | Inhibition of BET proteins | Phase 1/2 | Completed | 30% Stable disease | NCT02431260 |
| 43% Progressive disease | |||||
| SYHA1801 | BRD4 | Phase 1 | On going | Not reported | NCT04309968 |
| PLX51107 | BRD4 | Phase 1 | Completed | Tolerable except for patients with extensive hepatic metastasis | NCT04022785 |
| Phase 1 | On going | Not reported | NCT04022785 | ||
| BMS-986158 | Inhibition of BET proteins | Phase 1 | On going | Not reported | NCT03936465 |
| AZD5153 | BRD4 | Phase 1 | Completed | Tolerable | NCT03205176 |
| GSK525762 | Inhibition of BET proteins | Phase 1 | Completed | Manageable tolerability | NCT01587703 |
| Volasertib | PLK1 | Phase 3 | Completed | Did not meet primary objectif in combination with cytarabine | NCT01721876 |
| Onvansertib | PLK1 | Phase 2 | On going | Not reported | NCT03414034 |
| TAK-960 | PLK1 | Phase 1 | Completed | Terminated due to lack of efficacy | NCT01179399 |
| CYC140 | PLK1 | Phase 1 | Completed | Not reported | NCT03884829 |
| NMS-1286937 | PLK1 | Phase 1 | Completed | Tolerable | NCT01014429 |
| CX-4945 | CK2 | Phase 1/2 | Completed | Not reported | NCT02128282 |
| Phase 1/2 | On going | Not reported | NCT03904862 | ||
| MK-8776 | CHK1 | Phase 2 | Completed | Similar result as control arm | NCT01870596 |
| SRA737 | CHK1 | Phase 1/2 | Completed | Tolerable | NCT02797964 |
| Phase 1/2 | Completed | Tolerable | NCT02797977 | ||
| LY2603618 | CHK1 | Phase 1/2 | Completed | Toxicy when combined with pemetrexed+Cisplatin | NCT01139775 |
| Prexasertib | CHK1 | Phase 2 | On going | Tolerable, only modest activity in BRCA mutant HGSOC patients | NCT02203513 |
| Phase 2 | On going | Not reported | NCT03414047 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orhan, E.; Velazquez, C.; Tabet, I.; Sardet, C.; Theillet, C. Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy? Cancers 2021, 13, 2930. https://doi.org/10.3390/cancers13122930
Orhan E, Velazquez C, Tabet I, Sardet C, Theillet C. Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy? Cancers. 2021; 13(12):2930. https://doi.org/10.3390/cancers13122930
Chicago/Turabian StyleOrhan, Esin, Carolina Velazquez, Imene Tabet, Claude Sardet, and Charles Theillet. 2021. "Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy?" Cancers 13, no. 12: 2930. https://doi.org/10.3390/cancers13122930
APA StyleOrhan, E., Velazquez, C., Tabet, I., Sardet, C., & Theillet, C. (2021). Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy? Cancers, 13(12), 2930. https://doi.org/10.3390/cancers13122930