
Potential Biomarkers for Treatment Response to the BCL-2 Inhibitor Venetoclax: State of the Art and Future Directions



Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Basis of Venetoclax Activity
2.1. Apoptosis Activation and Control
2.2. Apoptosis Dysregulation in Hematolymphoid Neoplasms
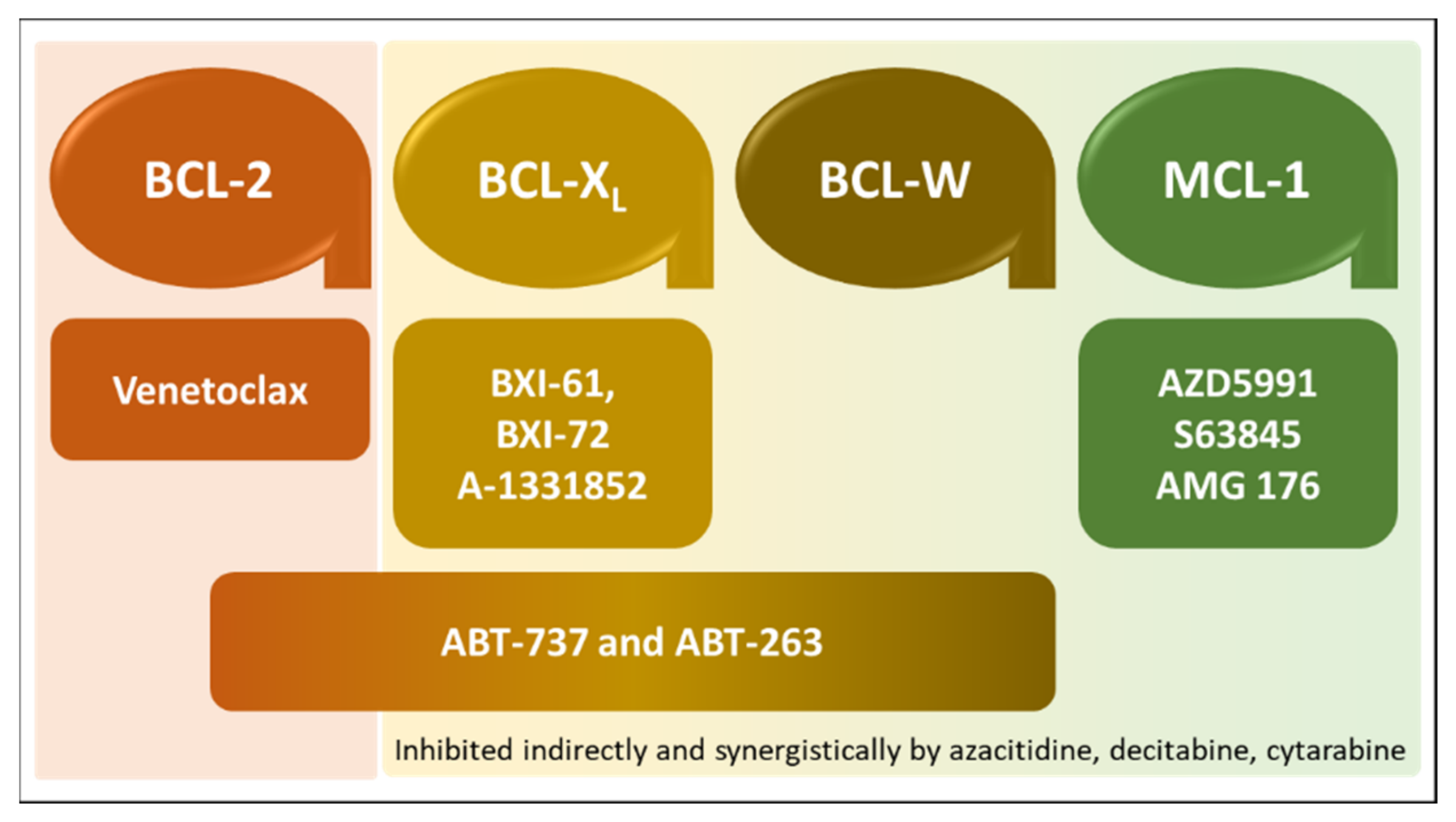
2.3. An Overview on BCL-2 Inhibition
2.4. Venetoclax and Its Clinical Utility
3. Mechanisms and Biomarkers of Resistance to Venetoclax
3.1. Mechanisms and Biomarkers of Venetoclax Response in CLL
3.1.1. BCL2 Mutations of Venetoclax Binding Site
3.1.2. Activation of Alternative Anti-Apoptotic Proteins and Pathways
3.1.3. BH3 Profiling as a BH3-Mimetic Drug Response Prediction Tool
3.1.4. Mutational Alterations Correlating with Venetoclax Response
3.1.5. Cytogenetic Alterations Correlating with Venetoclax Response
3.2. Mechanisms and Biomarkers of Venetoclax Response in AML
3.2.1. BCL2 Gene Alterations
3.2.2. Mutational Alterations Correlating with Venetoclax Response and Activation of Alternative Anti-Apoptosis Pathways
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Huang, D. Targeting BCL2 With BH3 Mimetics: Basic Science and Clinical Application of Venetoclax in Chronic Lymphocytic Leukemia and Related B Cell Malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef]
- Bose, P.; Gandhi, V.; Konopleva, M. Pathways and mechanisms of venetoclax resistance. Leuk. Lymphoma 2017, 58, 1–17. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the treatment paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Coutre, S.; Choi, M.; Furman, R.R.; Eradat, H.; Heffner, L.; Jones, J.A.; Chyla, B.; Zhou, L.; Agarwal, S.; Waskiewicz, T.; et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood 2018, 131, 1704–1711. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Konopleva, M.Y. A venetoclax bench-to-bedside story. Nature Cancer 2021, 2, 3–5. [Google Scholar] [CrossRef]
- Roberts, A.W.; Stilgenbauer, S.; Seymour, J.F.; Huang, D.C.S. Venetoclax in Patients with Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 4527–4533. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Tessoulin, B.; Papin, A.; Gomez-Bougie, P.; Bellanger, C.; Amiot, M.; Pellat-Deceunynck, C.; Chiron, D. BCL2-Family Dysregulation in B-Cell Malignancies: From Gene Expression Regulation to a Targeted Therapy Biomarker. Front. Oncol. 2018, 8, 645. [Google Scholar] [CrossRef]
- Rossi, D. Quest of biomarkers for venetoclax-treated CLL. Blood 2019, 134, 97–98. [Google Scholar] [CrossRef]
- Khoury, J.D. The evolving potential of companion diagnostics. Scand. J. Clin. Lab. Invest. Suppl. 2016, 245, S22–S25. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Kist, M.; Vucic, D. Cell death pathways: Intricate connections and disease implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Medeiros, L.J.; Rassidakis, G.Z.; McDonnell, T.J.; Abruzzo, L.V.; Lai, R. Expression of Mcl-1 in mantle cell lymphoma is associated with high-grade morphology, a high proliferative state, and p53 overexpression. J. Pathol. 2003, 199, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.F.; Yu, Q.Q.; Young, K.H. Critically dysregulated signaling pathways and clinical utility of the pathway biomarkers in lymphoid malignancies. Chronic Dis. Transl. Med. 2018, 4, 29–44. [Google Scholar] [CrossRef]
- Milani, M.; Byrne, D.P.; Greaves, G.; Butterworth, M.; Cohen, G.M.; Eyers, P.A.; Varadarajan, S. DRP-1 is required for BH3 mimetic-mediated mitochondrial fragmentation and apoptosis. Cell Death Dis. 2017, 8, e2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Gibson, C.J.; Davids, M.S. BCL-2 Antagonism to Target the Intrinsic Mitochondrial Pathway of Apoptosis. Clin. Cancer Res. 2015, 21, 5021–5029. [Google Scholar] [CrossRef] [Green Version]
- Marques-Piubelli, M.L.; Schlette, E.J.; Khoury, J.D.; Furqan, F.; Vega, F.; Soto, L.M.S.; Wistuba, I.I.; Wierda, W.G.; Konopleva, M.; Ferrajoli, A.; et al. Expression of BCL2 alternative proteins and association with outcome in CLL patients treated with venetoclax. Leuk. Lymphoma 2020, 62, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Banker, D.E.; Groudine, M.; Norwood, T.; Appelbaum, F.R. Measurement of spontaneous and therapeutic agent-induced apoptosis with BCL-2 protein expression in acute myeloid leukemia. Blood 1997, 89, 243–255. [Google Scholar] [CrossRef]
- Bensi, L.; Longo, R.; Vecchi, A.; Messora, C.; Garagnani, L.; Bernardi, S.; Tamassia, M.G.; Sacchi, S. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 1995, 80, 98–102. [Google Scholar]
- Huang, J.; Fairbrother, W.; Reed, J.C. Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev. Hematol. 2015, 8, 283–297. [Google Scholar] [CrossRef]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma 2014, 55, 1980–1992. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Bociek, R.G.; Pavletic, S.; et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2007, 25, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.B.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gribben, J.; Itri, L.M.; et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009, 27, 5208–5212. [Google Scholar] [CrossRef]
- Chanan-Khan, A.A.; Niesvizky, R.; Hohl, R.J.; Zimmerman, T.M.; Christiansen, N.P.; Schiller, G.J.; Callander, N.; Lister, J.; Oken, M.; Jagannath, S. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk. Lymphoma 2009, 50, 559–565. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.; Cohen, G.M. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br. J. Haematol. 2013, 163, 139–142. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, L.; Shen, K.; Jin, H.; Li, H.; Cheng, Y.; Ma, X. Efficacy and Safety of Bcl-2 Inhibitor Venetoclax in Hematological Malignancy: A Systematic Review and Meta-Analysis of Clinical Trials. Front. Pharmacol. 2019, 10, 697. [Google Scholar] [CrossRef]
- Lampson, B.L.; Davids, M.S. The Development and Current Use of BCL-2 Inhibitors for the Treatment of Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leverson, J.D.; Cojocari, D. Hematologic Tumor Cell Resistance to the BCL-2 Inhibitor Venetoclax: A Product of Its Microenvironment? Front. Oncol. 2018, 8, 458. [Google Scholar] [CrossRef] [Green Version]
- Carrington, E.M.; Zhan, Y.; Brady, J.L.; Zhang, J.G.; Sutherland, R.M.; Anstee, N.S.; Schenk, R.L.; Vikstrom, I.B.; Delconte, R.B.; Segal, D.; et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017, 24, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, S.J.F.; Davids, M.S. Breaking through BCL-2 inhibition in CLL. Blood 2020, 135, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.; Balakrishnan, S.; Fox, M.; Seedhouse, C.H.; Russell, N.H. Genetic biomarkers predict response to dual BCL-2 and MCL-1 targeting in acute myeloid leukaemia cells. Oncotarget 2018, 9, 37777–37789. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Moujalled, D.M.; Pomilio, G.; Ghiurau, C.; Ivey, A.; Salmon, J.; Rijal, S.; Macraild, S.; Zhang, L.; Teh, T.C.; Tiong, I.S.; et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2019, 33, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Dey, J.; Deckwerth, T.L.; Kerwin, W.S.; Casalini, J.R.; Merrell, A.J.; Grenley, M.O.; Burns, C.; Ditzler, S.H.; Dixon, C.P.; Beirne, E.; et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci. Rep. 2017, 7, 18007. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, S.; Ylanko, J.; Shi, Y.; Hariharan, S.; Oakes, C.C.; Brauer, P.M.; Zuniga-Pflucker, J.C.; Leber, B.; Spaner, D.E.; Andrews, D.W. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood 2016, 128, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef]
- Khoury, J.D.; Catenacci, D.V. Next-generation companion diagnostics: Promises, challenges, and solutions. Arch. Pathol. Lab. Med. 2015, 139, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef]
- Thijssen, R.; Slinger, E.; Weller, K.; Geest, C.R.; Beaumont, T.; van Oers, M.H.; Kater, A.P.; Eldering, E. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica 2015, 100, e302–e306. [Google Scholar] [CrossRef] [Green Version]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e313. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Blombery, P.; Thompson, E.R.; Nguyen, T.; Birkinshaw, R.W.; Gong, J.N.; Chen, X.; McBean, M.; Thijssen, R.; Conway, T.; Anderson, M.A.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, G.S.; Tat, T.T.; Misra, S.; Hill, B.T.; Smith, M.R.; Almasan, A.; Mazumder, S. Cyclin E/Cdk2-dependent phosphorylation of Mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget 2015, 6, 16912–16925. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P.; Ruvolo, V.R.; Benton, C.B.; AlRawi, A.; Burks, J.K.; Schober, W.; Rolke, J.; Tidmarsh, G.; Hail, N., Jr.; Davis, R.E.; et al. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim. Biophys. Acta 2016, 1863, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Alford, S.E.; Kothari, A.; Loeff, F.C.; Eichhorn, J.M.; Sakurikar, N.; Goselink, H.M.; Saylors, R.L.; Jedema, I.; Falkenburg, J.H.; Chambers, T.C. BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 2015, 75, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Song, T.; Chai, G.; Liu, Y.; Yu, X.; Wang, Z.; Zhang, Z. Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br. J. Pharmacol. 2016, 173, 471–483. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, M.; Pettitt, A.; Varadarajan, S.; Cohen, G.M. BH3 profiling and a toolkit of BH3-mimetic drugs predict anti-apoptotic dependence of cancer cells. Br. J. Cancer 2016, 114, 638–641. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Deng, J.; Wiestner, A.; Lannutti, B.J.; Wang, L.; Wu, C.J.; Wilson, W.H.; Brown, J.R.; Letai, A. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood 2012, 120, 3501–3509. [Google Scholar] [CrossRef] [Green Version]
- Pallis, M.; Burrows, F.; Ryan, J.; Grundy, M.; Seedhouse, C.; Abdul-Aziz, A.; Montero, J.; Letai, A.; Russell, N. Complementary dynamic BH3 profiles predict co-operativity between the multi-kinase inhibitor TG02 and the BH3 mimetic ABT-199 in acute myeloid leukaemia cells. Oncotarget 2017, 8, 16220–16232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderquist, R.S.; Crawford, L.; Liu, E.; Lu, M.; Agarwal, A.; Anderson, G.R.; Lin, K.H.; Winter, P.S.; Cakir, M.; Wood, K.C. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 2018, 9, 3513. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Ma, S.; Kipps, T.J.; Coutre, S.E.; Davids, M.S.; Eichhorst, B.; Hallek, M.; Byrd, J.C.; Humphrey, K.; Zhou, L.; et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood 2019, 134, 111–122. [Google Scholar] [CrossRef]
- Anderson, M.A.; Tam, C.; Lew, T.E.; Juneja, S.; Juneja, M.; Westerman, D.; Wall, M.; Lade, S.; Gorelik, A.; Huang, D.C.S.; et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood 2017, 129, 3362–3370. [Google Scholar] [CrossRef] [Green Version]
- Mato, A.R.; Thompson, M.; Allan, J.N.; Brander, D.M.; Pagel, J.M.; Ujjani, C.S.; Hill, B.T.; Lamanna, N.; Lansigan, F.; Jacobs, R.; et al. Real-world outcomes and management strategies for venetoclax-treated chronic lymphocytic leukemia patients in the United States. Haematologica 2018, 103, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Blanco, G.; Espinet, B. Genetic abnormalities in chronic lymphocytic leukemia: Where we are and where we go. Biomed. Res. Int. 2014, 2014, 435983. [Google Scholar] [CrossRef]
- Pospisilova, S.; Sutton, L.A.; Malcikova, J.; Tausch, E.; Rossi, D.; Montserrat, E.; Moreno, C.; Stamatopoulos, K.; Gaidano, G.; Rosenquist, R.; et al. Innovation in the prognostication of chronic lymphocytic leukemia: How far beyond TP53 gene analysis can we go? Haematologica 2016, 101, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qian, J.; Wang, H.; Wang, Y.; Zhang, Y.; Qian, P.; Lou, Y.; Jin, J.; Zhu, H. Not BCL2 mutation but dominant mutation conversation contributed to acquired venetoclax resistance in acute myeloid leukemia. Biomark Res. 2021, 9, 30. [Google Scholar] [CrossRef]
- Zhang, H.; Wilmot, B.; Bottomly, D.; Kurtz, S.E.; Eide, C.A.; Damnernsawad, A.; Romine, K.; Patel, S.; Druker, B.J.; Mcweeney, S.K.; et al. Biomarkers Predicting Venetoclax Sensitivity and Strategies for Venetoclax Combination Treatment. Blood 2018, 132, 175. [Google Scholar] [CrossRef]
- Zhang, H.; Nakauchi, Y.; Kohnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Cantero Aguilar, L.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 inhibition with venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Wang, G.; Wang, Y.; Caldwell, J.T.; Edwards, H.; Xie, C.; Taub, J.W.; Li, C.; Lin, H.; Ge, Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014, 28, 1557–1560. [Google Scholar] [CrossRef]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 17, E202–E205. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Wierenga, A.T.; van den Heuvel, F.A.; Woldhuis, R.R.; Kluit, D.S.; Jaques, J.; Schuringa, J.J.; Vellenga, E. Elevated VMP1 expression in acute myeloid leukemia amplifies autophagy and is protective against venetoclax-induced apoptosis. Cell Death Dis. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef]
- Campos, E.D.V.; Pinto, R. Targeted therapy with a selective BCL-2 inhibitor in older patients with acute myeloid leukemia. Hematol. Transfus. Cell Ther. 2019, 41, 169–177. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huemer, F.; Melchardt, T.; Jansko, B.; Wahida, A.; Jilg, S.; Jost, P.J.; Klieser, E.; Steiger, K.; Magnes, T.; Pleyer, L.; et al. Durable remissions with venetoclax monotherapy in secondary AML refractory to hypomethylating agents and high expression of BCL-2 and/or BIM. Eur. J. Haematol. 2019, 102, 437–441. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 12, 791–803. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Pathways of Resistance | Reported Examples of Mechanisms of Resistance | Type of Malignancy | Type of Study | Reference |
|---|---|---|---|---|
| BCL2 mutations of venetoclax-binding site | Gly101Val mutation * | CLL | Patients derived samples | [51] |
| Phe104Leu/Cys mutations | DLBCL, FL, MCL, and leukemia cell lines | In vitro (preclinical) | [52] | |
| Alternative anti-apoptotic proteins pathways | Overexpression of BCL-XL, MCL-1, and BFL-1. | CLL | Patients derived samples | [53] |
| Amp1q leading to MCL-1 overexpression | CLL | In vitro (preclinical) followed by patients derived samples | [54] | |
| Kinase-mediated survival signals leading to BCL-XL, MCL-1, and A1 | CLL | Patients derived samples | [47] | |
| MCL-1 overexpression | AML | In vitro (preclinical) | [42] | |
| MCL-1 overexpression | AML | In vitro (preclinical) | [43] | |
| PI3K/mTOR pathway and BCL-2 → MCL-1 overexpression | AML | In vitro (preclinical) | [45] | |
| Cyclin-dependent kinase 9 inhibition (CDK-9) → MCL-1 overexpression | DLBCL | In vitro (preclinical) | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salah, H.T.; DiNardo, C.D.; Konopleva, M.; Khoury, J.D. Potential Biomarkers for Treatment Response to the BCL-2 Inhibitor Venetoclax: State of the Art and Future Directions. Cancers 2021, 13, 2974. https://doi.org/10.3390/cancers13122974
Salah HT, DiNardo CD, Konopleva M, Khoury JD. Potential Biomarkers for Treatment Response to the BCL-2 Inhibitor Venetoclax: State of the Art and Future Directions. Cancers. 2021; 13(12):2974. https://doi.org/10.3390/cancers13122974
Chicago/Turabian StyleSalah, Haneen T., Courtney D. DiNardo, Marina Konopleva, and Joseph D. Khoury. 2021. "Potential Biomarkers for Treatment Response to the BCL-2 Inhibitor Venetoclax: State of the Art and Future Directions" Cancers 13, no. 12: 2974. https://doi.org/10.3390/cancers13122974