Simple Summary
Altered regulation of the cell cycle is a hallmark of cancer. The recent clinical success of the inhibitors of CDK4 and CDK6 has convincingly demonstrated that targeting cell cycle components may represent an effective anti-cancer strategy, at least in some cancer types. However, possible applications of CDK4/6 inhibitors in patients with ovarian cancer is still under evaluation. Here, we describe the possible biological role of CDK4 and CDK6 complexes in ovarian cancer and provide the rationale for the use of CDK4/6 inhibitors in this pathology, alone or in combination with other drugs. This review, coupling basic, preclinical and clinical research studies, could be of great translational value for investigators attempting to design new clinical trials for the better management of ovarian cancer patients.
Abstract
Alterations in components of the cell-cycle machinery are present in essentially all tumor types. In particular, molecular alterations resulting in dysregulation of the G1 to S phase transition have been observed in almost all human tumors, including ovarian cancer. These alterations have been identified as potential therapeutic targets in several cancer types, thereby stimulating the development of small molecule inhibitors of the cyclin dependent kinases. Among these, CDK4 and CDK6 inhibitors confirmed in clinical trials that CDKs might indeed represent valid therapeutic targets in, at least some, types of cancer. CDK4 and CDK6 inhibitors are now used in clinic for the treatment of patients with estrogen receptor positive metastatic breast cancer and their clinical use is being tested in many other cancer types, alone or in combination with other agents. Here, we review the role of CDK4 and CDK6 complexes in ovarian cancer and propose the possible use of their inhibitors in the treatment of ovarian cancer patients with different types and stages of disease.
1. Introduction
In 2000, Hanahan and Weinberg, by putting together the principal advancements of cancer research during the previous 25 years, provided the scientific community with the knowledge of what, at that time, were the hallmarks of cancer [1]. An update of these hallmarks was provided by the same authors in 2011 [2]. The sustained proliferative signaling and the evasion from growth suppression were and remained two of the most typical traits reported in virtually all cancer cells. Both these features are tightly related to an altered regulation of the cell cycle progression. Ten years later, we know that a dysregulated cell cycle is not only a hallmark of cancer, but also one of its main vulnerabilities, targetable by specific biological therapies.
Here, we will focus on the possible roles that the inhibition of two central regulators of cell cycle progression, namely CDK4 and CDK6, may play in the context of ovarian cancer. We will highlight how they have been used so far for the treatment of this disease and what are the future steps that need to be made to possibly expand their success.
2. The Role of CDK4 and CDK6 in the Control of Cell Cycle Progression
Cell cycle progression is a tightly controlled process that ensures the correct division of one cell into two daughter cells with the same genetic material [3]. To this aim, a series of enzymatic chain reactions ensures that, after a mitogenic stimuli, a cell may first enter the cell cycle from a status of quiescence, faithfully replicate the DNA, and, finally, segregate the DNA content in the two daughter cells [3].
Three principal families of proteins govern the progression through the mitotic cell cycle: cyclins, cyclin dependent kinases (CDK) and CDK inhibitors (CDKI). The consecutive activation of different CDKs allows the cell exit from a state of quiescence, termed G0, and the entering in the cell cycle in a phase known as G1 (or Gap 1) during which the cell prepares to duplicate its genetic content. This happens during the S (synthesis) phase of the cell cycle that follows the G1 phase and precedes the phase G2 (or Gap 2), in which the cell prepares to divide during mitosis (M phase). Each transition from one phase to the other is regulated by the activation of specific cyclins-CDKs complexes. The complexes containing the D type of cyclins (i.e., D1, D2 and D3) and the CDK 4 and 6 are the first complexes activated by the cell following a mitogenic signal. Their activation is necessary for the cells to exit from the state of quiescence, proceed along the G1 phase and bypass the so-called restriction point, a checkpoint that, once bypassed, commits the cell to complete the mitotic cycle and divide. It is therefore not surprising that all cell cycle proteins involved in the regulation of G1 phase progression are often deregulated in cancer and have been for long time conceived as possible therapeutic targets.
In a state of quiescence or of cell cycle resting (phase G0 or early G1), the CDK4/6 are inactive, bound to the CDKI of the INK4 family (i.e., p15INK4B, p16INK4A, p18INK4C and p19INK4D), and the expression of cyclin Ds very low or absent. Contextually, CDK2, the other CDK involved in the G1 to S phase transition, is maintained in an inactive state bound to the CDKI, p27KIP1. Under these conditions, the tumor suppressor Rb (Retinoblastoma protein) is active and binds and represses the transcription factors of the E2F family that, in turn, are necessary to transcribe pro-proliferative genes. Upon mitogenic stimuli (e.g., growth factor stimulation of tyrosine kinase receptors, estrogen receptor activation, etc.), cyclins Ds are rapidly transcribed and p27KIP1 is tyrosine phosphorylated by nonreceptor tyrosine kinases, such as Src family members [4,5]. These events allow the formation of an active CDK4/6-cyclin D-p27KIP1 complex that translocates in the nucleus and phosphorylates Rb, causing the de-repression of E2F family members. As a consequence, E2F factors transcribe the genes necessary to form an active cyclin E/CDK2 complex that allows bypassing the restriction point and, eventually, commits the cell to complete the division.
It is therefore clear that even a small dysregulation of the INK4, CDK4/6, cyclins D and p27KIP1 proteins may profoundly impact on the physiological control of the cell cycle. Accordingly, a great body of evidence has demonstrated that one or more of these genes are altered in most human cancers and their alteration often predicts poorer patients’ prognosis.
A clear input toward the comprehension of the role of these proteins in human cancers comes from the generation and characterization of genetically modified mice, knock out (KO) for one or more of these genes. The description of these models is out of the scope of this review but, as a whole, the picture depicted by these studies has defined some pillars and highlighted several important issues. First, the role of cyclin D1, D2 and D3 seems to be organ specific, with cyclin D1 mostly involved in the development of the mammary gland and the retina [6], cyclin D2 in the development of the gonads [7] and cyclin D3 in the maturation of T lymphocytes [8]. Mice deficient for Cdk4 are viable and only display proliferative defects in specific endocrine cell types and the same is true for Cdk6 KO mice, which develop normally with only minor hematopoietic defects [9]. Interestingly, mice lacking both Cdk4 and Cdk6 die at the end of gestation, due to severe anemia, although these embryos display normal organogenesis and most cell types seem to proliferate normally [9]. Very similar results were obtained by the analysis of mice null for the three cyclin Ds [10]. These results indicate that CDK4 and CDK6 have many overlapping functions and that they are not essential for cell cycle entry, since most normal cells are able to activate alternative mechanisms to initiate cell proliferation. Further, these studies suggest that inhibition of CDK4 and CDK6 could be exploited to specifically control the proliferation of tumor cells that rely on their activity.
This hypothesis has been experimentally validated in mice, demonstrating, for instance, that mice lacking cyclin D1, or Cdk4, or mice expressing kinase-deficient cyclin D1-Cdk complexes were resistant to HER2- or Ras-driven mammary tumorigenesis, but not to the one sustained by c-Myc overexpression [11,12]. Accordingly, pharmacological inhibition of Cdk4 and Cdk6 prevented tumor development in mouse models of breast cancer, sustained by cyclin D1, and leukemia, sustained by cyclin D3 [13].
These seminal studies have open the way to the testing of specific CDK4 and CDK6 small molecule inhibitors (hereafter CDK4/6i) to treat human tumors, in particular estrogen receptor positive (ER+) breast cancers, in which their use, combined with hormonal therapies, has proved highly effective in the control of disease progression [14,15,16,17].
3. Non-Cell Cycle Dependent Activities of CDK4 and CDK6
Beyond their common activity in governing G1-S cell cycle progression, CDK4 and CDK6 also display kinase dependent and independent functions, therefore regulating many other cellular processes like DNA damage and repair, transcription, senescence, invasion, metabolism and immune response. The discovery of these roles of CDK4 and CDK6 in several cellular processes has certainly positively contributed to the further clinical development of CDK4/6i in different types of cancer, used alone or in combination with other therapeutic approaches [14,18,19,20,21,22,23]. In particular, accumulating evidences indicate that CDK4 and CDK6 play a central and originally unanticipated role in the regulation of the DNA Damage Response (DDR). For instance, it has been shown that CDK4/6 inhibition combined with ionizing radiation results in a shift from homologous recombination (HR) to error-prone non-homologous end-joining (NHEJ) DNA repair mechanism in pancreatic adenocarcinoma, ER+ BC and TNBC models [24,25,26]. Similarly, CDK6 expression protects epithelial ovarian cancer cells from platinum-induced cell death controlling ATR transcription through FOXO3a phosphorylation and stabilization, directly impacting on DDR during the S phase of the cell cycle [27].
More recently, transcriptome analyses performed in different cancer cell models modified for CDK4 and CDK6 expression or activity, highlighted that, beyond the control of the G1 phase of the cell cycle, CDK4 and CDK6 might play non-cell cycle related functions via the regulation of transcription [28]. In particular, it was reported that while CDK4 controls pro-metastatic inflammatory pathways, CDK6 mainly regulates DNA damage, repair and replication, through the modulation of genes, such as TK1, POLD3, POLE2, CENPI and DTL, essential for these processes [28]. Similarly, Watt and colleagues observed that breast cancer cell lines and PDX models treated with the CDK4/6i abemaciclib or palbociclib, displayed a remodeling of the chromatin architecture with a wide activation of transcription enhancers and super-enhancers. These transcriptional modifications are involved in the regulation of luminal differentiation, apoptosis and immune response [29]. Whether these activities of CDK4/6i are due to unexpected off target effects of the inhibitors or effectively to the inhibition of CDK4 and/or CDK6 kinases activities, although largely expected, has not been formally demonstrated [29].
Regulation of gene transcription by CDK4 and CDK6 has a well-established role in the regulation of senescence, through the phosphorylation of the transcription factor FOXM1 and described as an important event in melanoma and ovarian cancer [30,31]. FOXM1 has been identified as a CDK4 and CDK6 substrate using an unbiased phospho-proteomic screening. This approach also highlighted that CDK4- and CDK6-specific target proteins exist [30]. Among others, FOXO3a and several splicing factors seems to be CDK6-specific substrates, supporting the hypothesis that this kinase might regulate not only gene transcription, but also DDR and RNA splicing [27,30].
CDK4 and CDK6 were also associated with the regulation of protein ubiquitination and stability. In particular, it has been demonstrated that both these CDKs can bind and phosphorylate the deubiquitinase USP51 and DUB3, to control ZEB1 [32] and SNAIL1 [33] expression, respectively, thereby modulating the metastatic phenotype of cancer cells. Interestingly, ZEB1 is also a direct phosphorylation target of CDK6 [27,30], suggesting that multiple layers of control exist between CDK4/6 activity and the regulation of cell invasion. Accordingly, it has been shown that the CDK4-cyclin D1 complex could control cell adhesion to the Extra-Cellular Matrix (ECM) and cell motility by directly phosphorylating the Paxillin-Rac1 complex in the membrane ruffles [34].
Intriguing evidences also suggest that CDK4/6 activity is implicated in cell metabolism, as demonstrated by the increase in oxidative phosphorylation when the Rb pathway is inhibited in pancreatic cancers [35]. At mechanistic level, CDK4/6i lead to the compensatory activation of the MEK and mTOR pathways. Accordingly, combined pharmacological inhibition of CDK4/6 and MEK potentiated the cytostatic effect of CDK4/6i, while the use of CDK4/6i plus mTOR pathway inhibitors increased cell death [35]. These observations might have high translational relevance, since both MEK and mTOR pathway inhibitors are currently used in clinic for the treatment of several types of human cancers.
Last but not least, evidences accumulated in the last few years highlighted a role for CDK4 and CDK6 inhibition in triggering anti-tumor immunity. The first evidences were obtained in breast cancer with abemaciclib, suggesting that suppression of the Rb–E2F axis leads to a reduced expression of the methyl-transferase DNMT1 and, thus, to the hypomethylation of genes that regulate the immune response. As a consequence, CDK4/6i increased antigen presentation by tumor cells and reduced tumor infiltration by immunosuppressive regulatory T cells [36]. These observations support the possibility that the combined use of CDK4/6i and immune checkpoint inhibitors (e.g., anti-PD-L1 antibodies) may result in synergistic antitumor activity [36,37]. Interestingly, it has been also demonstrated that the cyclin D/CDK4 complex might regulate PD-L1 protein abundance, leading to its proteasomal degradation and that CDK4/6i increases PD-L1 levels in vivo, in breast cancer models [38]. Relevant to this review, these observations were recently confirmed in ovarian cancer models, in which treatment with abemaciclib increases both immune infiltration and the activity of cytotoxic lymphocytes, making ovarian cancer more sensitive to the PD-1 blockade [39].
4. Mechanism of Action of CDK4/6 Inhibitors
Targeting the CDK activity as an anticancer strategy has been tested since the late 90s, when the toxicity profile of a pan-CDK inhibitor (flavopiridol) was tested in 76 patients with refractory malignancies [40]. Yet, the way of administration (i.e., intravenous injection), the low therapeutic index and the high toxicity profiles, at the concentrations necessary to inhibit their targets, as already observed for other pan-CDK inhibitors, such as roscovitine, cooled the enthusiasm for this type of targeted therapies [41]. Similarly, the second generation of more selective and more potent CDK inhibitors, such as dinaciclib, demonstrated little clinical activity in several cancer types [41].
In 2001, the first of a kind CDK4/6 specific inhibitor (i.e., PD0332991, then renamed palbociclib) was developed as an orally available pyrido [2,3-d]pyrimidines derivate and proved the ability to block cancer cells in the G1 phase of the cell cycle [42,43]. This small molecule inhibitor is an ATP-competitive inhibitor of CDK4/6-cyclin D, inhibiting these complexes with exquisite selectivity, 20-fold higher than CDK2/cyclin E complex and more than 100-fold higher than FGFR [42,43].
Then, experiments in cells and mouse models of cancer confirmed that PD0332991 displayed very promising antitumor activities [13,43,44], but it took many years until its therapeutic value became really appreciated, as described by Garber in 2014 [45].
The unexpected and impressive activity reported for palbociclib in breast cancer patients when used in combination with anti-estrogen therapy [45] then rapidly stimulated the design of other CDK4/6 inhibitors that rapidly entered in clinical development. Three orally available CDK4/6 inhibitors are currently approved for the treatment of patients with metastatic breast cancer (i.e., palbociclib, ribociclib and abemaciclib) [41] and one, administered intravenously and with shorter half-life, has been developed to prevent chemotherapy-induced myelosuppression [46]. Many others are under clinical development and have been recently reviewed elsewhere [47]. Here, we will focus on the activity of the orally available compounds that have been shown to bind both the monomeric CDK4/6 kinase or the CDK4/6-cyclin D complexes and, by binding the CDK4/6 in the cleft between the N-terminal and C-terminal lobes, inhibit the ATP binding [48]. An important distinctive feature is that abemaciclib, compared to palbociclib and ribociclib, displays a wider spectrum of action and inhibits at nanomolar concentration not only CDK4 and CDK6, but also CDK9 (Table 1).

Table 1.
Activity of orally available CDK4/6 inhibitors on cyclin/CDKs complexes, in vitro.
According to the pivotal role of CDK4 and CDK6 in driving cell cycle progression through the G1 phase of the cell cycle, the most prominent effect of all CDK4/6i is the block of cell proliferation [41]. Biochemically, this cell cycle blockage is accompanied by the inhibition of Rb phosphorylation, an event that can be observed both in vitro and in vivo [41,49]. Therefore, it has been postulated that tumors with alteration in proteins regulating the progression through the G1 phase of the cell cycle will be the most sensitive to these inhibitors. Deletion of Rb family genes and/or amplification of CDK4, CDK6 or cyclin Ds have been proposed as relevant markers of resistance to these drugs [41,47]. However, in line with the multiple activities of CDK4 and CDK6, it is highly expected that the use and the success of CDK4/6i will be also related to many other alterations, present both in tumor cells and in tumor microenvironment.
5. Expression of CDK4 and CDK6 Containing Complexes in Ovarian Cancer
Cell cycle proteins are deregulated in almost all types of human cancers, including epithelial ovarian carcinomas [41]. Interestingly, it has been observed that in High Grade Serous Ovarian Cancer (HGSOC) amplification of cyclin E1 (CCNE1) is associated with resistance to platinum-based chemotherapies [50], suggesting that targeting the cell cycle in these patients could represent an effective therapeutic approach.
Here, we have reviewed the studies reporting the expression of the principal regulators of the G1 to S phase transition of the cell cycle. Considering these data as whole, we found that a significant fraction of ovarian cancers that have been analyzed display an aberrant expression of cyclins, CDKs and/or CDKI (Table 2), supporting the hypothesis that these tumors could be potentially sensitive to CDK4/6i.
Table 2.
Expression of cell cycle proteins in ovarian cancer.
Slamon and colleagues firstly tried to identify which ovarian cancer type may be sensitive to the CDK4/6i [85]. For this aim, they screened 40 ovarian cancer cell lines for their sensitivity to palbociclib and identified the so-called Rb1-proficient cell lines, with low p16INK4A and CCNE1 expression, as the most responsive to CDK4/6i. They next analyzed the expression of other regulators of CDK4 and CDK6 signaling, including the CDKIs p15INK4B, p18INK4C, p19INK4D, p21WAF1 and p27KIP1, the Rb family of proteins, Rb2/p130 and Rb3/p107, the D-cyclins (D1, D2 and D3) and the E2F1 transcription factor. However, none of the above proteins strongly correlated with in vitro sensitivity to palbociclib. Conversely, they found an association between cyclin D1 gene (CCND1) amplification and resistance to palbociclib [85]. The analysis of the expression of Rb1 and p16INK4A in a panel of 263 epithelial ovarian cancer samples, by immunohistochemistry (IHC), demonstrated that low/null expression of p16INK4A and Rb1 were associated with shorter patients’ progression free survival [85] and suggested that these tumors could be the ones most benefitting from the treatment with CDK4/6i, alone or in combination with chemotherapy.
We and others have evaluated the expression of both CDK4 and CDK6 in epithelial ovarian cancers with interesting results. In general, it was observed that CDK6, more than CDK4, was expressed at high levels in epithelial ovarian cancer [27,55,76,77,78]. Many evidences suggest that CDK6 regulates the sensitivity to platinum in ovarian cancer cells and that its high expression is associated with a platinum-resistant phenotype [27,77]. Interestingly, CDK6 was mostly localized in the cytoplasm although this localization does not correlate with patients’ survival [78]. In line with this finding, data from publicly available gene expression profile (GEP) datasets suggest that high CDK6, but not CDK4, mRNA expression predicts shorter progression free survival in ovarian cancer patients [27]. Similar observation has been made for D type cyclins, with a clear difference between cyclin D1 (no prognostic role) and cyclin D3, whose high expression predicts shorter patients’ overall/progression free survival (OS/PFS) [27]. However, the results on mRNA expression were not always confirmed by studies that looked at protein expression (Table 2), suggesting that the significance of the expression of D type cyclins in ovarian cancer should be better evaluated. It is interesting to note that cyclin D2, which is a FSH responding gene necessary for gonadal cell proliferation [7], seems to be more and/or exclusively expressed in ovarian germ cell tumors [59,60].
Almost all reported studies suggest that CCNE1 amplification and overexpression predicts shorter patients’ survival (Table 2), in line with the original observation that it is associated with resistance to platinum-based therapy in HGSOC [50].
From studies that analyzed CDKI expression in ovarian cancer samples, it appears clear that the CDKN1A gene (p16INKA) had the most relevant prognostic significance. More than one study points to p16INK4A expression as a biomarker of longer patients’ survival (Table 2). Interesting results were obtained for both CDKN1A (p21WAF1) and CDKN1B (p27KIP1), whose expression was generally associated with lower grade and better patients’ outcome (Table 2). Interestingly, p21WAF1 expression seems to be restricted to tumors with wild type TP53 gene [80] and loss of p27KIP1 expression has been reported to be an early event in the development of HGSOC [93]. Therefore, it is likely that in HGSOC, in which the first event during transformation is usually the acquisition of a mutation of TP53 gene, both these CDKI are inactivated, eventually leading to higher activity of CDK complexes.
The expression of the others members of the INK4 family of CDKI (CDKN2B, C and D) has been less studied in ovarian cancer (Table 2) but it has been surprisingly observed that CDKN2D (p19INK4D) expression was associated with higher tumor stage and shorter patients’ survival [92], which might deserve further investigation in future studies.
6. Combination Strategies of CDK4/6 Inhibitors with Conventional Cytotoxic Agents
Most of chemotherapeutics currently used as anticancer agents exert their main effect by targeting actively proliferating cells. In particular, platinum salts, anthracyclines and topoisomerase inhibitors are mostly active during the S phase of the cell cycle, while DNA is duplicated, whereas taxanes, vinca alkaloids and eribulin mostly target the mitotic process during the M phase. Accordingly, the side effects of these conventional cytotoxic agents are particularly evident in healthy cells that are highly proliferating in organs and tissues, such as the bone marrow, the epithelial tissues of the gastrointestinal tract and the skin.
Since CDK4/6i block both normal and cancer cells in the G0/G1 phase of the cell cycle, it was firstly hypothesized that the use of these novel agents in combination with standard chemotherapies could be detrimental for the efficacy of the cancer treatment [94]. Nevertheless, a growing body of preclinical evidence now supports the sequential use of standard chemotherapeutics followed by CDK4/6 inhibitors [95] and, as consequence, early phase clinical trials testing these combinations are thriving. This drastic route change is mainly due to the recent discoveries regarding the role played by CDK4/6 in DDR. These new data in fact suggest that CDK4/6 inhibitors may well cooperate with DNA or mitotic damaging agents to enhance their anti-tumor activity, if used with the proper time schedule [47].
In ovarian cancer, platinum salts are the most commonly used and active agents for patients with newly diagnosed disease, as well as for those with a platinum-sensitive recurrent disease [96]. It is well known that platinum induces the formation of DNA single and double strand breaks, leading to the activation of DDR. It is also recognized that the proper activation of this cellular response, in which the ATR kinase plays a central role, often represents a cause of escape from platinum-induced apoptosis [97,98]. Notably, recent studies from our lab have demonstrated that CDK6, mainly in complex with cyclin D3, contributes to the activation of DDR in HGSOC and induces the expression of ATR [27]. As expectable, the activation of this survival mechanism eventually protects TP53 deficient ovarian cancer cells from platinum-induced cell death. Accordingly, CDK6 inhibition with palbociclib significantly increased platinum-induced apoptosis both in vitro and in vivo [27]. Similarly, it was reported that ribociclib showed synergism with platinum, in xenograft models of HGSOC exposed to concurrent ribociclib and cisplatin treatment, followed by maintenance with ribociclib [99]. On the basis of these promising preclinical results, the use of CDK4/6i to increase the platinum efficacy in ovarian cancer patients is currently being tested in several clinical trials (Table 3).
Table 3.
Ongoing clinical trials with CDK4/6 inhibitors in ovarian cancer patients.
Based on recent studies and in line with what just mentioned above, it is also possible that inhibition of CDK4/6 may have synergistic activity with inhibition of Poly ADP-ribose polymerase (PARPi). Since PARPi increases genomic instability in cancer cells and CDK4/6i impair the DDR, this strategy could induce HR deficiency, even in HR proficient ovarian cancers. Recent evidences in preclinical ovarian cancer models, combining palbociclib with the PARPi olaparib, have indeed demonstrated a clear synergism in ovarian cancers with high Myc expression [100]. The clinical synergism between PARPi and CDK4/6i is now under clinical investigation in different cancer settings (Table 3).
7. Use of CDK4/6 Inhibitors as Single Agents in Ovarian Cancer Patients
The effectiveness of the CDK4/6 inhibitors as single agent (palbociclib) was explored in a phase II trial in heavily pretreated ovarian cancer patients. Efficacy was assessed in 30 patients, mainly serous ovarian cancer patients, showing 9/30 (30%) patients that were progression-free at 6 months. The authors reported one partial response and 17 disease stabilizations, with a median PFS of 3.7 months. Palbociclib was well tolerated, and hematological toxicities of grade 3 and 4 were reported in only 10 patients [101]. Based on the preclinical data collected by the same group, the expression of Rb1, CDKN2A and/or CCNE1, along with the amplification of CCND1, could represent good biomarkers to predict response [85]. These biomarkers are absolutely necessary, since the activity of palbociclib in an unselected population seems to be modest, at least when it is administered as single agent.
The association between CDK4/6i with anti-hormonal therapies certainly deserves to be mentioned for its promising results in gynecological malignancy, particularly in hormone receptor positive endometrial and ovarian cancers where endocrine agents are generally used alone in later treatment lines. A trial combining ribociclib and letrozole in estrogen receptor positive (>10%) endometrial and ovarian cancer patients demonstrated promising clinical activity in relapsed ovarian cancer patients [102]. About 50% of the 40 patients enrolled were progression-free after 12 weeks and, interestingly, the greatest benefit was seen in low grade serous ovarian cancer (LGSOC) patients, who had PFS longer than 24 months. However, since only 3 LGSOC patients were enrolled in the trial, these data will need further confirmation [102].
8. Clinical Experiences on the Use of CDK 4/6i with Chemotherapy
The combination of platinum-based chemotherapy with CDK4/6i has been tested in several solid malignancies. The study by Swiecicki et al. was of particular interest because the combination was specifically tested to improve the efficacy of platinum [103]. This phase II trial enrolled recurrent and metastatic head and neck cancer patients, treated with carboplatin on day 1 (with a starting dose of 5AUC) and palbociclib (125 mg daily) on days 1–14, every three weeks. This schedule of carboplatin and palbociclib showed disappointing antitumor activity, with a 12-week disease control rate of 33% (5/18 stable disease and 1/18 partial response), a median PFS of 2.9 months and significant associated toxicities: grade 3 or higher toxicities were seen in 79% of patients, with the most common being myelosuppression [103]. It is conceivable that this weak anti-tumor activity is due, at least in part, to an incorrect timing of palbociclib administration: given in concomitance with platinum and not sequentially, palbociclib may not be able to prevent the recovery from cytotoxic DNA damage. In conclusion, the treatment schedule proposed by the Swiecicki et al. was suboptimal not only for the lack of a synergistic effect between the two drugs, but also for the high rate of toxicity that could have mined dose intensity. Best dose/sequence finding studies are greatly needed to overcome these limitations.
In another study, the CDK4/6i trilaciclib was used in combination with chemotherapy in breast cancer patients with metastatic triple negative disease, in order to reduce dose-limiting hematological and myeloid-toxicities of chemotherapy [104]. Intriguingly, despite the fact that addition of trilaciclib failed to protect immune cells and bone marrow from chemotherapy-induced damage, the arms in which patients received trilaciclib showed a clinically meaningful survival advantage compared to the arm treated with chemotherapy alone (median OS was 12.6 months with chemotherapy alone, 20 months and 17.8 months in the two trilaciclib arms) [104]. Major limitations of the trial were the small sample size and the open label design. Nevertheless, the possibility that this difference in survival could be linked to a synergistic effect of chemotherapies with the CDK4/6i certainly deserves further investigation.
An intermitting schedule of palbociclib followed by paclitaxel was evaluated in a phase 1 study in breast cancer patients [24]. The trial stemmed from promising preclinical results and tested the possibility to enhance treatment efficacy by using intermittent, alternating dosing with palbociclib and paclitaxel, in 27 metastatic Rb1-proficient breast cancer patients [105]. The recommended dose and schedule found was 75 mg of palbociclib administered for 3 consecutive days, started at least 24 h after each dose of weekly paclitaxel. The clinical benefit rate was 55% at the recommended schedule and it was observed across all receptor subtypes. Thanks to these results, the association of palbociclib with paclitaxel is currently under investigation also in a phase 1 trial, in patients with advanced pancreatic cancer (NCT02501902).
9. CDK4/6 Inhibitors Safety and Tolerability Profile
Pivotal phase III trials in advanced breast cancer, where CDK4/6 inhibitors were administered in combination with endocrine therapy (e.g., aromatase inhibitor, fulvestrant), have revealed that palbociclib, ribociclib and abemaciclib were generally well tolerated. In these trials, as well as in clinical practice, adverse events were easily manageable with treatment delay, dose modification and supportive care measures.
Regarding class-specific side effects, which are commonly seen with all three inhibitors, hematological toxicities, in particular neutropenia, are most frequent with palbociclib and ribociclib than abemaciclib.
Regarding drug-specific side effects associated with different CDK4/6 inhibitors, ribociclib might cause hepatotoxicity and has also been linked with reversible prolongation of the QT interval. On the other hand, abemaciclib can induce diarrhea more frequently than palbociclib and ribociclib and in the MONARCH studies venous thromboembolic events were found more frequently in the abemaciclib than in the placebo group. Finally, since all three molecules are substrates of the CYP3A4 enzyme, concomitant therapies with moderate or strong inhibitors or inducers of CYP3A4 must be avoided.
10. Conclusions and Future Perspectives
CDK4/6i have proven to be highly effective in the treatment of advanced stage estrogen receptor-positive HER2-negative breast cancer and, in these patients, the combination of CDK4/6i and hormonal therapy represents now the standard of care [106].
Based on this evident clinical benefit, the activity of CDK4/6i as single agent or in combination therapies is being tested also in other types of cancer.
Ovarian cancer represents a complex and extremely heterogeneous group of neoplasms. Based on accumulating preclinical and clinical evidences, the use of CDK4/6i could be successfully tested in several settings. At this regard, it is to note that CDK4/6 signaling is frequently altered in these tumors, although, depending on the histotype considered, the specific altered gene may vary (see above). Keeping this concept in mind and if they will be tested appropriately, looking at the different disease grades, stages, histologies and molecular alterations, we expect that the use of CDK4/6i will be of great promise also for ovarian cancer patients (Figure 1).
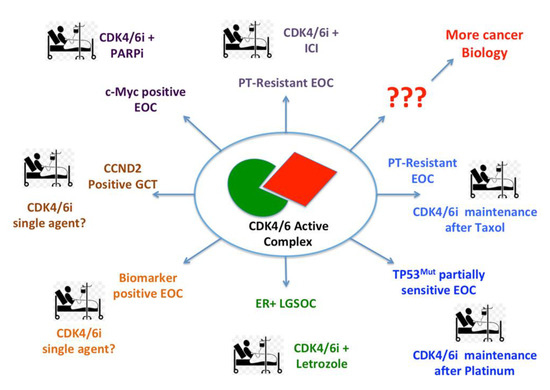
Figure 1.
Possible therapeutic approaches based on the use of CDK4/6i in ovarian cancer patients.
Accumulating preclinical and clinical evidences suggest that CDK4/6i could be used alone and in association with other chemo- or targeted-therapies in different tumor contexts. Here, we propose possible applications for ovarian cancer patients with different types/stages of disease (see text for detail). EOC = Epithelial Ovarian Cancer; GCT = Germ Cell Tumors; LGSOC = Low Grade Serous Ovarian Cancer; PARPi = PARPs (Poly ADP Ribose Polymerases) inhibitors; ICI = Immune Checkpoint Inhibitors; PT = Platinum; ER+ = Estrogen Receptor positive.
The use of CDK4/6i as single agents in ovarian cancer patients does not seem to hold promise and is probably not worth pursuing further, unless specific biomarkers of activity will be identified. At this regard, based on the biology of germ cell tumors (GCT) and on the fact that they usually overexpress cyclin D2, it could be worth testing if CDK4/6i might be particularly active in these histotypes. Indeed palbociclib seems to be active in a subset of patients with GCT [107] and novel in vitro experiments support a dual role for CDK4/6i in controlling GCT cell proliferation and survival [108]. However, since these rare germinal tumors usually affect adolescents or young adults, the set-up of a clinical trial will be particularly complicated. On the other hand, for the same reason, it would be particularly relevant sparing whenever possible the adverse effects of chemotherapy to these young patients.
The results of the phase II trial, testing the efficacy of ribociclib and letrozole in patients with recurrent ovarian cancer and showing a high response rate in the few patients with low grade tumors, absolutely merit to be further explored, broadening the cohort of LGSOC patients.
In HGSOC, which almost invariably carry mutations in the tumor suppressor gene TP53 and, for this reason, are predicted to be highly sensitive to ATR inhibition [27,109], it would be worth testing the association between CDK4/6i and cisplatin, using CDKi as maintenance therapy. This schedule should, in principle, allow the strongest synergistic effect and avoid the accumulation of hematological toxicities, as observed in head and neck patients that were concomitantly treated with carboplatin and palbociclib [103]. This regimen could be promising also in recurrent ovarian cancer patients that have been treated with platinum-based therapy and still display partial response to platinum. In platinum resistant disease, based on the recent evidences obtained in pancreatic cancer models [25], it would be worth testing the sequential administration of taxanes followed by CDK4/6i as maintenance therapy.
In the setting of ovarian cancer patients with c-Myc overexpressing tumors it would be important testing if the association of CDK4/6i with PARPi is really effective, as demonstrated in preclinical models [100]. This regimen would have the advantage of being a chemo-free approach, particularly indicated for frail patients.
No mature evidence currently supports the adoption of immunotherapy in ovarian cancer patients [110]. Since CDK4/6i can profoundly impact on the immune response to different cancer types, it would be interesting to verify if the use of CDK4/6i may improve the efficacy of immune-checkpoint inhibitors, which would be particularly needed for platinum resistant patients that still have very few valid therapeutic options.
From all the studies conducted so far, we can undoubtedly conclude that the design and management of these novel clinical trials will only be possible in the frame of large, cooperative, translational, multidisciplinary groups, dedicated to the research and the care of gynecological tumors, of which ovarian cancers are not the most frequent but certainly still the most deadly ones.
Author Contributions
Conceptualization, G.B.; writing—original draft preparation, A.D., M.B., N.M.-K., G.B.; writing—review and editing, R.S., F.P., B.B., G.B.; supervision, G.B.; funding acquisition, G.B., B.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Ministero della Salute (RF-2016-02361040; CO-2018-12367051 and Ricerca Corrente core grant), Alleanza Contro il Cancro (RCR-2019-23669115), Ministero degli Esteri e Cooperazione Internazionale (PGR01230), Regione Autonoma Friuli Venezia Giulia (TICHEP grant) and 5x1000 CRO, to G.B.; from Associazione Italiana Ricerca sul Cancro (AIRC) (IG #20061), to B.B.
Acknowledgments
We like to thank all members of the SCICC lab for fruitful scientific discussion. We apologize to all colleagues whose work, for space limitations, was not discussed or cited.
Conflicts of Interest
The authors declare no conflict of interest related to this work.
Abbreviations
CCC: Clear Cell Carcinoma; CISH: Chromogenic In Situ Hybridization; EOC: Epithelial Ovarian Cancer; EC: Endometrioid Carcinoma; FISH: Fluorescent In Situ Hybridization; GEP: Gene Expression Profile; GCT: Granulosa Cell Tumors; HGSOC: High Grade Serous Ovarian Carcinoma; IHC: Immunohistochemistry; LGSOC: Low Grade Serous Ovarian Carcinoma; MS-PCR: Methylation-Specific Polymerase Chain Reaction; MC: Mucinous Carcinoma; NOSa: Not Otherwise Specified adenocarcinoma; PSF: Progression Free Survival; OS: Overall Survival; RPPA: Reverse Phase Protein Array; STIC: Serous Tubal Intraepithelial Carcinoma.
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P. A Long Twentieth Century of the Cell Cycle and Beyond. Cell 2000, 100, 71–78. [Google Scholar] [CrossRef]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilensek, Z.; Keidel, E.-M.; Waddell, M.B.; Jäkel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-Inhibitory Activity and Stability of P27Kip1 Are Directly Regulated by Oncogenic Tyrosine Kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.; Sun, J.; Arnaout, A.; Kahn, H.; Hanna, W.; Narod, S.; Sun, P.; Tan, C.-K.; Hengst, L.; Slingerland, J. P27 Phosphorylation by Src Regulates Inhibition of Cyclin E-Cdk2. Cell 2007, 128, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Donaher, J.L.; Parker, S.B.; Li, T.; Fazeli, A.; Gardner, H.; Haslam, S.Z.; Bronson, R.T.; Elledge, S.J.; Weinberg, R.A. Cyclin D1 Provides a Link between Development and Oncogenesis in the Retina and Breast. Cell 1995, 82, 621–630. [Google Scholar] [CrossRef]
- Sicinski, P.; Donaher, J.L.; Geng, Y.; Parker, S.B.; Gardner, H.; Park, M.Y.; Robker, R.L.; Richards, J.S.; McGinnis, L.K.; Biggers, J.D.; et al. Cyclin D2 Is an FSH-Responsive Gene Involved in Gonadal Cell Proliferation and Oncogenesis. Nature 1996, 384, 470–474. [Google Scholar] [CrossRef]
- Sicinska, E.; Aifantis, I.; Le Cam, L.; Swat, W.; Borowski, C.; Yu, Q.; Ferrando, A.A.; Levin, S.D.; Geng, Y.; Von Boehmer, H.; et al. Requirement for Cyclin D3 in Lymphocyte Development and T Cell Leukemias. Cancer Cell 2003, 4, 451–461. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef]
- Kozar, K.; Ciemerych, M.A.; Rebel, V.I.; Shigematsu, H.; Zagozdzon, A.; Sicinska, E.; Geng, Y.; Yu, Q.; Bhattacharya, S.; Bronson, R.T.; et al. Mouse Development and Cell Proliferation in the Absence of D-Cyclins. Cell 2004, 118, 477–491. [Google Scholar] [CrossRef]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific Protection against Breast Cancers by Cyclin D1 Ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef]
- Yu, Q.; Sicinska, E.; Geng, Y.; Ahnström, M.; Zagozdzon, A.; Kong, Y.; Gardner, H.; Kiyokawa, H.; Harris, L.N.; Stål, O.; et al. Requirement for CDK4 Kinase Function in Breast Cancer. Cancer Cell 2006, 9, 23–32. [Google Scholar] [CrossRef]
- Choi, Y.J.; Li, X.; Hydbring, P.; Sanda, T.; Stefano, J.; Christie, A.L.; Signoretti, S.; Look, A.T.; Kung, A.L.; Von Boehmer, H.; et al. The Requirement for Cyclin D Function in Tumor Maintenance. Cancer Cell 2012, 22, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Giudici, F.; Giuliano, M.; Cristofanilli, M.; Arpino, G.; Del Mastro, L.; Puglisi, F.; De Placido, S.; Paris, I.; De Placido, P.; et al. Overall Survival of CDK4/6-Inhibitor-Based Treatments in Clinically Relevant Subgroups of Metastatic Breast Cancer: Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2020, 112, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.; Gerratana, L.; Corvaja, C.; Pelizzari, G.; Franceschin, G.; Bertoli, E.; Palmero, L.; Zara, D.; Alberti, M.; Buriolla, S.; et al. First- and Second-Line Treatment Strategies for Hormone-Receptor (HR)-Positive HER2-Negative Metastatic Breast Cancer: A Real-World Study. Breast 2021, 57, 104–112. [Google Scholar] [CrossRef]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Niesvizky, R.; Badros, A.Z.; Costa, L.J.; Ely, S.A.; Singhal, S.B.; Stadtmauer, E.A.; Haideri, N.A.; Yacoub, A.; Hess, G.; Lentzsch, S.; et al. Phase 1/2 Study of Cyclin-Dependent Kinase (CDK)4/6 Inhibitor Palbociclib (PD-0332991) with Bortezomib and Dexamethasone in Relapsed/Refractory Multiple Myeloma. Leuk. Lymphoma 2015, 56, 3320–3328. [Google Scholar] [CrossRef]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 Trial of Palbociclib in Adult Patients with Recurrent RB1-Positive Glioblastoma. J. Neurooncol. 2018, 140, 477–483. [Google Scholar] [CrossRef]
- Martin, P.; Ruan, J.; Furman, R.; Rutherford, S.; Allan, J.; Chen, Z.; Huang, X.; DiLiberto, M.; Chen-Kiang, S.; Leonard, J.P. A Phase I Trial of Palbociclib plus Bortezomib in Previously Treated Mantle Cell Lymphoma. Leuk. Lymphoma 2019, 60, 2917–2921. [Google Scholar] [CrossRef]
- Besse, B.; Barlesi, F.; Demedts, I.; Fuentes Pradera, J.; Robinet, G.; Gazzah, A.; Soldatenkova, V.; Frimodt-Moller, B.; Kim, J.S.; Vansteenkiste, J. A Phase 1b Study of Necitumumab in Combination with Abemaciclib in Patients with Stage IV Non-Small Cell Lung Cancer. Lung Cancer 2019, 137, 136–143. [Google Scholar] [CrossRef]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and Cetuximab in Platinum-Resistant and in Cetuximab-Resistant Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicentre, Multigroup, Phase 2 Trial. Lancet Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA Damage Response by Therapeutic CDK4/6 Inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Pesch, A.M.; Hirsh, N.H.; Chandler, B.C.; Michmerhuizen, A.R.; Ritter, C.L.; Androsiglio, M.P.; Wilder-Romans, K.; Liu, M.; Gersch, C.L.; Larios, J.M.; et al. Short-Term CDK4/6 Inhibition Radiosensitizes Estrogen Receptor-Positive Breast Cancers. Clin. Cancer Res. 2020, 26, 6568–6580. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, A.; Sonego, M.; Pellizzari, I.; Pellarin, I.; Canzonieri, V.; D’Andrea, S.; Benevol, S.; Sorio, R.; Giorda, G.; Califano, D.; et al. CDK6 Protects Epithelial Ovarian Cancer from Platinum-Induced Death via FOXO3 Regulation. EMBO Mol. Med. 2017, 9, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Boudreault, J.; Wang, N.; Poulet, S.; Daliah, G.; Yan, G.; Moamer, A.; Burgos, S.A.; Sabri, S.; Ali, S.; et al. Differential Regulation of Cancer Progression by CDK4/6 Plays a Central Role in DNA Replication and Repair Pathways. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Watt, A.C.; Cejas, P.; DeCristo, M.J.; Metzger-Filho, O.; Lam, E.Y.N.; Qiu, X.; BrinJones, H.; Kesten, N.; Coulson, R.; Font-Tello, A.; et al. CDK4/6 Inhibition Reprograms the Breast Cancer Enhancer Landscape by Stimulating AP-1 Transcriptional Activity. Nat. Cancer 2021, 2, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef]
- Liu, G.; Sun, Y.; Ji, P.; Li, X.; Cogdell, D.; Yang, D.; Parker Kerrigan, B.C.; Shmulevich, I.; Chen, K.; Sood, A.K.; et al. MiR-506 Suppresses Proliferation and Induces Senescence by Directly Targeting the CDK4/6-FOXM1 Axis in Ovarian Cancer. J. Pathol. 2014, 233, 308–318. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Ou, Y.; Yang, G.; Deng, K.; Wang, Q.; Wang, Z.; Wang, W.; Zhang, Q.; Wang, H.; et al. CDK4/6 Inhibition Blocks Cancer Metastasis through a USP51-ZEB1-Dependent Deubiquitination Mechanism. Signal Transduct. Target. Ther. 2020, 5, 25. [Google Scholar] [CrossRef]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T.; et al. CDK4/6-Dependent Activation of DUB3 Regulates Cancer Metastasis through SNAIL1. Nat. Commun. 2017, 8, 13923. [Google Scholar] [CrossRef]
- Fusté, N.P.; Fernández-Hernández, R.; Cemeli, T.; Mirantes, C.; Pedraza, N.; Rafel, M.; Torres-Rosell, J.; Colomina, N.; Ferrezuelo, F.; Dolcet, X.; et al. Cytoplasmic Cyclin D1 Regulates Cell Invasion and Metastasis through the Phosphorylation of Paxillin. Nat. Commun. 2016, 7, 11581. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 Kinase Destabilizes PD-L1 via Cullin 3-SPOP to Control Cancer Immune Surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Zhang, Q.-F.; Li, J.; Jiang, K.; Wang, R.; Ge, J.-L.; Yang, H.; Liu, S.-J.; Jia, L.-T.; Wang, L.; Chen, B.-L. CDK4/6 Inhibition Promotes Immune Infiltration in Ovarian Cancer and Synergizes with PD-1 Blockade in a B Cell-Dependent Manner. Theranostics 2020, 10, 10619–10633. [Google Scholar] [CrossRef]
- Senderowicz, A.M.; Headlee, D.; Stinson, S.F.; Lush, R.M.; Kalil, N.; Villalba, L.; Hill, K.; Steinberg, S.M.; Figg, W.D.; Tompkins, A.; et al. Phase I Trial of Continuous Infusion Flavopiridol, a Novel Cyclin-Dependent Kinase Inhibitor, in Patients with Refractory Neoplasms. J. Clin. Oncol. 1998, 16, 2986–2999. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Toogood, P.L. Cyclin-Dependent Kinase Inhibitors for Treating Cancer. Med. Res. Rev. 2001, 21, 487–498. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef] [PubMed]
- Baughn, L.B.; Di Liberto, M.; Wu, K.; Toogood, P.L.; Louie, T.; Gottschalk, R.; Niesvizky, R.; Cho, H.; Ely, S.; Moore, M.A.S.; et al. A Novel Orally Active Small Molecule Potently Induces G1 Arrest in Primary Myeloma Cells and Prevents Tumor Growth by Specific Inhibition of Cyclin-Dependent Kinase 4/6. Cancer Res. 2006, 66, 7661–7667. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. The Cancer Drug That Almost Wasn’t. Science 2014, 345, 865–867. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-White, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; Van Hoogdalem, E.-J.; et al. Transient CDK4/6 Inhibition Protects Hematopoietic Stem Cells from Chemotherapy-Induced Exhaustion. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Worsley, S.D.; Jennings, B.A.; Khalil, K.H.; Mole, M.; Girling, A.C. Cyclin D1 Amplification and Expression in Human Breast Carcinoma: Correlation with Histological Prognostic Markers and Oestrogen Receptor Expression. Clin. Mol. Pathol. 1996, 49, M46–M50. [Google Scholar] [CrossRef] [PubMed]
- Dhar, K.K.; Branigan, K.; Parkes, J.; Howells, R.E.; Hand, P.; Musgrove, C.; Strange, R.C.; Fryer, A.A.; Redman, C.W.; Hoban, P.R. Expression and Subcellular Localization of Cyclin D1 Protein in Epithelial Ovarian Tumour Cells. Br. J. Cancer 1999, 81, 1174–1181. [Google Scholar] [CrossRef]
- Sui, L.; Tokuda, M.; Ohno, M.; Hatase, O.; Hando, T. The Concurrent Expression of P27(Kip1) and Cyclin D1 in Epithelial Ovarian Tumors. Gynecol. Oncol. 1999, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Bali, A.; O’Brien, P.M.; Edwards, L.S.; Sutherland, R.L.; Hacker, N.F.; Henshall, S.M. Cyclin D1, P53, and P21Waf1/Cip1 Expression Is Predictive of Poor Clinical Outcome in Serous Epithelial Ovarian Cancer. Clin. Cancer Res. 2004, 10, 5168–5177. [Google Scholar] [CrossRef]
- Masciullo, V.; Scambia, G.; Marone, M.; Giannitelli, C.; Ferrandina, G.; Bellacosa, A.; Benedetti Panici, P.; Mancuso, S. Altered Expression of Cyclin D1 and CDK4 Genes in Ovarian Carcinomas. Int. J. Cancer 1997, 74, 390–395. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yanaihara, N.; Okamoto, A.; Nikaido, T.; Saito, M.; Takakura, S.; Yasuda, M.; Sasaki, H.; Ochiai, K.; Tanaka, T. Cyclin D1 Predicts the Prognosis of Advanced Serous Ovarian Cancer. Exp. Ther. Med. 2011, 2, 213–219. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Wang, H.; Makki, M.S.; Wen, J.; Dai, Y.; Shi, Q.; Liu, Q.; Zhou, X.; Wang, J. Overexpression of β-Catenin and CyclinD1 Predicts a Poor Prognosis in Ovarian Serous Carcinomas. Int. J. Clin. Exp. Pathol. 2014, 7, 264–271. [Google Scholar]
- Abdelrahman, A.E.; Fathy, A.; Elsebai, E.A.; Nawar, N.; Etman, W.M. Prognostic Impact of Apaf-1, Cyclin D1, and AQP-5 in Serous Ovarian Carcinoma Treated with the First-Line Chemotherapy. Ann. Diagn. Pathol. 2018, 35, 27–37. [Google Scholar] [CrossRef]
- Chu, S.; Rushdi, S.; Zumpe, E.T.; Mamers, P.; Healy, D.L.; Jobling, T.; Burger, H.G.; Fuller, P.J. FSH-Regulated Gene Expression Profiles in Ovarian Tumours and Normal Ovaries. Mol. Hum. Reprod. 2002, 8, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kyrönlahti, A.; Rämö, M.; Tamminen, M.; Unkila-Kallio, L.; Butzow, R.; Leminen, A.; Nemer, M.; Rahman, N.; Huhtaniemi, I.; Heikinheimo, M.; et al. GATA-4 Regulates Bcl-2 Expression in Ovarian Granulosa Cell Tumors. Endocrinology 2008, 149, 5635–5642. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sakuma, M.; Akahira, J.-I.; Ito, K.; Niikura, H.; Moriya, T.; Okamura, K.; Sasano, H.; Yaegashi, N. Promoter Methylation Status of the Cyclin D2 Gene Is Associated with Poor Prognosis in Human Epithelial Ovarian Cancer. Cancer Sci. 2007, 98, 380–386. [Google Scholar] [CrossRef]
- Chang, L.; Guo, R.; Yuan, Z.; Shi, H.; Zhang, D. LncRNA HOTAIR Regulates CCND1 and CCND2 Expression by Sponging MiR-206 in Ovarian Cancer. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 1289–1303. [Google Scholar] [CrossRef]
- Levidou, G.; Korkolopoulou, P.; Thymara, I.; Vassilopoulos, I.; Saetta, A.A.; Gakiopoulou, H.; Konstantinidou, A.; Kairi-Vassilatou, E.; Pavlakis, K.; Patsouris, E. Expression and Prognostic Significance of Cyclin D3 in Ovarian Adenocarcinomas. Int. J. Gynecol. Pathol. 2007, 26, 410–417. [Google Scholar] [CrossRef]
- Sui, L.; Dong, Y.; Ohno, M.; Sugimoto, K.; Tai, Y.; Hando, T.; Tokuda, M. Implication of Malignancy and Prognosis of P27(Kip1), Cyclin E, and Cdk2 Expression in Epithelial Ovarian Tumors. Gynecol. Oncol. 2001, 83, 56–63. [Google Scholar] [CrossRef]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene Amplification CCNE1 Is Related to Poor Survival and Potential Therapeutic Target in Ovarian Cancer. Cancer 2010, 116, 2621–2634. [Google Scholar] [CrossRef]
- Pils, D.; Bachmayr-Heyda, A.; Auer, K.; Svoboda, M.; Auner, V.; Hager, G.; Obermayr, E.; Reiner, A.; Reinthaller, A.; Speiser, P.; et al. Cyclin E1 (CCNE1) as Independent Positive Prognostic Factor in Advanced Stage Serous Ovarian Cancer Patients—A Study of the OVCAD Consortium. Eur. J. Cancer 2014, 50, 99–110. [Google Scholar] [CrossRef]
- Yang, L.; Fang, D.; Chen, H.; Lu, Y.; Dong, Z.; Ding, H.-F.; Jing, Q.; Su, S.-B.; Huang, S. Cyclin-Dependent Kinase 2 Is an Ideal Target for Ovary Tumors with Elevated Cyclin E1 Expression. Oncotarget 2015, 6, 20801–20812. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.L.; Drapkin, R. Cyclin E1 Deregulation Occurs Early in Secretory Cell Transformation to Promote Formation of Fallopian Tube-Derived High-Grade Serous Ovarian Cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef]
- Kuhn, E.; Wang, T.-L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shih, I.-M. CCNE1 Amplification and Centrosome Number Abnormality in Serous Tubal Intraepithelial Carcinoma: Further Evidence Supporting Its Role as a Precursor of Ovarian High-Grade Serous Carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Kuhn, E.; Wu, R.-C.; Ogawa, H.; Bahadirli-Talbott, A.; Mao, T.-L.; Sugimura, H.; Shih, I.-M.; Wang, T.-L. CCNE1 Copy-Number Gain and Overexpression Identify Ovarian Clear Cell Carcinoma with a Poor Prognosis. Mod. Pathol. 2017, 30, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Aziz, D.; Etemadmoghadam, D.; Caldon, C.E.; Au-Yeung, G.; Deng, N.; Hutchinson, R.; Australian Ovarian Cancer Study Group; Bowtell, D.; Waring, P. 19q12 Amplified and Non-Amplified Subsets of High Grade Serous Ovarian Cancer with Overexpression of Cyclin E1 Differ in Their Molecular Drivers and Clinical Outcomes. Gynecol. Oncol. 2018, 151, 327–336. [Google Scholar] [CrossRef]
- Sapoznik, S.; Aviel-Ronen, S.; Bahar-Shany, K.; Zadok, O.; Levanon, K. CCNE1 Expression in High Grade Serous Carcinoma Does Not Correlate with Chemoresistance. Oncotarget 2017, 8, 62240–62247. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Wilson, A.J.; Hirst, J.; Roby, K.F.; Fadare, O.; Crispens, M.A.; Beeghly-Fadiel, A.; Khabele, D. CCNE1 and BRD4 Co-Amplification in High-Grade Serous Ovarian Cancer Is Associated with Poor Clinical Outcomes. Gynecol. Oncol. 2020, 157, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.M.; Enwere, E.; McIntyre, J.B.; Wilson, H.; Nwaroh, C.; Wiebe, N.; Ou, Y.; Liu, S.; Wiedemeyer, K.; Rambau, P.F.; et al. Combined CCNE1 High-Level Amplification and Overexpression Is Associated with Unfavourable Outcome in Tubo-Ovarian High-Grade Serous Carcinoma. J. Pathol. Clin. Res. 2020, 6, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Marone, M.; Scambia, G.; Giannitelli, C.; Ferrandina, G.; Masciullo, V.; Bellacosa, A.; Benedetti-Panici, P.; Mancuso, S. Analysis of Cyclin E and CDK2 in Ovarian Cancer: Gene Amplification and RNA Overexpression. Int. J. Cancer 1998, 75, 34–39. [Google Scholar] [CrossRef]
- Sui, L.; Dong, Y.; Ohno, M.; Goto, M.; Inohara, T.; Sugimoto, K.; Tai, Y.; Hando, T.; Tokuda, M. Inverse Expression of Cdk4 and P16 in Epithelial Ovarian Tumors. Gynecol. Oncol. 2000, 79, 230–237. [Google Scholar] [CrossRef]
- Duan, L.; Yan, Y.; Wang, G.; Xing, Y.L.; Sun, J.; Wang, L.L. ΜiR-182-5p Functions as a Tumor Suppressor to Sensitize Human Ovarian Cancer Cells to Cisplatin through Direct Targeting the Cyclin Dependent Kinase 6 (CDK6). J. BUON 2020, 25, 2279–2286. [Google Scholar] [PubMed]
- Perrone, F.; Baldassarre, G.; Indraccolo, S.; Signoriello, S.; Chiappetta, G.; Esposito, F.; Ferrandina, G.; Franco, R.; Mezzanzanica, D.; Sonego, M.; et al. Biomarker Analysis of the MITO2 Phase III Trial of First-Line Treatment in Ovarian Cancer: Predictive Value of DNA-PK and Phosphorylated ACC. Oncotarget 2016, 7, 72654–72661. [Google Scholar] [CrossRef][Green Version]
- Anttila, M.A.; Kosma, V.M.; Hongxiu, J.; Puolakka, J.; Juhola, M.; Saarikoski, S.; Syrjänen, K. P21/WAF1 Expression as Related to P53, Cell Proliferation and Prognosis in Epithelial Ovarian Cancer. Br. J. Cancer 1999, 79, 1870–1878. [Google Scholar] [CrossRef]
- Schmider, A.; Gee, C.; Friedmann, W.; Lukas, J.J.; Press, M.F.; Lichtenegger, W.; Reles, A. P21 (WAF1/CIP1) Protein Expression Is Associated with Prolonged Survival but Not with P53 Expression in Epithelial Ovarian Carcinoma. Gynecol. Oncol. 2000, 77, 237–242. [Google Scholar] [CrossRef]
- Rose, S.L.; Goodheart, M.J.; DeYoung, B.R.; Smith, B.J.; Buller, R.E. P21 Expression Predicts Outcome in P53-Null Ovarian Carcinoma. Clin. Cancer Res. 2003, 9, 1028–1032. [Google Scholar] [PubMed]
- Skirnisdottir, I.; Seidal, T. Association of P21, P21 P27 and P21 P53 Status to Histological Subtypes and Prognosis in Low-Stage Epithelial Ovarian Cancer. Cancer Genom. Proteom. 2013, 10, 27–34. [Google Scholar]
- Alrehaili, A.A.; AlMourgi, M.; Gharib, A.F.; Elsawy, W.H.; Ismail, K.A.; Hagag, H.M.; Anjum, F.; Raafat, N. Clinical Significance of P27 Kip1 Expression in Advanced Ovarian Cancer. Appl. Cancer Res. 2020, 40, 6. [Google Scholar] [CrossRef]
- Hafez, M.M.; Alhoshani, A.R.; Al-Hosaini, K.A.; Alsharari, S.D.; Al Rejaie, S.S.; Sayed-Ahmed, M.M.; Al-Shabanah, O.A. SKP2/P27Kip1 Pathway Is Associated with Advanced Ovarian Cancer in Saudi Patients. Asian Pac. J. Cancer Prev. 2015, 16, 5807–5815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Konecny, G.E.; Winterhoff, B.; Kolarova, T.; Qi, J.; Manivong, K.; Dering, J.; Yang, G.; Chalukya, M.; Wang, H.-J.; Anderson, L.; et al. Expression of P16 and Retinoblastoma Determines Response to CDK4/6 Inhibition in Ovarian Cancer. Clin. Cancer Res. 2011, 17, 1591–1602. [Google Scholar] [CrossRef]
- Sallum, L.F.; Andrade, L.; Ramalho, S.; Ferracini, A.C.; de Andrade Natal, R.; Brito, A.B.C.; Sarian, L.O.; Derchain, S. WT1, P53 and P16 Expression in the Diagnosis of Low- and High-Grade Serous Ovarian Carcinomas and Their Relation to Prognosis. Oncotarget 2018, 9, 15818–15827. [Google Scholar] [CrossRef]
- Dong, Y.; Walsh, M.D.; McGuckin, M.A.; Gabrielli, B.G.; Cummings, M.C.; Wright, R.G.; Hurst, T.; Khoo, S.K.; Parsons, P.G. Increased Expression of Cyclin-Dependent Kinase Inhibitor 2 (CDKN2A) Gene Product P16INK4A in Ovarian Cancer Is Associated with Progression and Unfavourable Prognosis. Int. J. Cancer 1997, 74, 57–63. [Google Scholar] [CrossRef]
- Kommoss, S.; du Bois, A.; Ridder, R.; Trunk, M.J.; Schmidt, D.; Pfisterer, J.; Kommoss, F. AGO-OVAR Independent Prognostic Significance of Cell Cycle Regulator Proteins P16(INK4a) and PRb in Advanced-Stage Ovarian Carcinoma Including Optimally Debulked Patients: A Translational Research Subprotocol of a Randomised Study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group. Br. J. Cancer 2007, 96, 306–313. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, L.-E.; Wang, L.; Lu, K.H.; Mills, G.B.; Bondy, M.L.; Wei, Q. Methylation and Messenger RNA Expression of P15INK4b but Not P16INK4a Are Independent Risk Factors for Ovarian Cancer. Clin. Cancer Res. 2005, 11, 4968–4976. [Google Scholar] [CrossRef]
- Ozdemir, F.; Altinisik, J.; Karateke, A.; Coksuer, H.; Buyru, N. Methylation of Tumor Suppressor Genes in Ovarian Cancer. Exp. Ther. Med. 2012, 4, 1092–1096. [Google Scholar] [CrossRef]
- Arcellana-Panlilio, M.Y.; Egeler, R.M.; Ujack, E.; Magliocco, A.; Stuart, G.C.E.; Robbins, S.M.; Coppes, M.J. Evidence of a Role for the INK4 Family of Cyclin-Dependent Kinase Inhibitors in Ovarian Granulosa Cell Tumors. Genes Chromosom. Cancer 2002, 35, 176–181. [Google Scholar] [CrossRef]
- Felisiak-Golabek, A.; Dansonka-Mieszkowska, A.; Rzepecka, I.K.; Szafron, L.; Kwiatkowska, E.; Konopka, B.; Podgorska, A.; Rembiszewska, A.; Kupryjanczyk, J. P19(INK4d) MRNA and Protein Expression as New Prognostic Factors in Ovarian Cancer Patients. Cancer Biol. Ther. 2013, 14, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Levanon, K.; Duraisamy, S.; Liu, J.F.; Hirsch, M.S.; Hecht, J.L.; Drapkin, R. Stathmin 1, a Marker of PI3K Pathway Activation and Regulator of Microtubule Dynamics, Is Expressed in Early Pelvic Serous Carcinomas. Gynecol. Oncol. 2011, 123, 5–12. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.-K.; Perou, C.M.; Sharpless, N.E. Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Roberts, P.J.; Kumarasamy, V.; Witkiewicz, A.K.; Knudsen, E.S. Chemotherapy and CDK4/6 Inhibitors: Unexpected Bedfellows. Mol. Cancer Ther. 2020, 19, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial Ovarian Cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems Biology of Cisplatin Resistance: Past, Present and Future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Iyengar, M.; O’Hayer, P.; Cole, A.; Sebastian, T.; Yang, K.; Coffman, L.; Buckanovich, R.J. CDK4/6 Inhibition as Maintenance and Combination Therapy for High Grade Serous Ovarian Cancer. Oncotarget 2018, 9, 15658–15672. [Google Scholar] [CrossRef]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC Status as a Determinant of Synergistic Response to Olaparib and Palbociclib in Ovarian Cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wahner Hendrickson, A.E.; Jatoi, A.; Burton, J.K.; Paroly, J.; Glaspy, J.A.; Dowdy, S.C.; Slamon, D.J. A Multicenter Open-Label Phase II Study of the Efficacy and Safety of Palbociclib a Cyclin-Dependent Kinases 4 and 6 Inhibitor in Patients with Recurrent Ovarian Cancer. J. Clin. Oncol. 2016, 34, 5557. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Zanfagnin, V.; Hou, X.; Foster, N.R.; Asmus, E.J.; Wahner Hendrickson, A.; Jatoi, A.; Block, M.S.; Langstraat, C.L.; Glaser, G.E.; et al. Phase II Trial of Ribociclib and Letrozole in Patients with Relapsed Oestrogen Receptor-Positive Ovarian or Endometrial Cancers. ESMO Open 2020, 5, e000926. [Google Scholar] [CrossRef] [PubMed]
- Swiecicki, P.L.; Durm, G.; Bellile, E.; Bhangale, A.; Brenner, J.C.; Worden, F.P. A Multi-Center Phase II Trial Evaluating the Efficacy of Palbociclib in Combination with Carboplatin for the Treatment of Unresectable Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Investig. New Drugs 2020, 38, 1550–1558. [Google Scholar] [CrossRef]
- Tan, A.R.; Wright, G.S.; Thummala, A.R.; Danso, M.A.; Popovic, L.; Pluard, T.J.; Han, H.S.; Vojnović, Ž.; Vasev, N.; Ma, L.; et al. Trilaciclib plus Chemotherapy versus Chemotherapy Alone in Patients with Metastatic Triple-Negative Breast Cancer: A Multicentre, Randomised, Open-Label, Phase 2 Trial. Lancet Oncol. 2019, 20, 1587–1601. [Google Scholar] [CrossRef]
- Clark, A.S.; McAndrew, N.P.; Troxel, A.; Feldman, M.; Lal, P.; Rosen, M.; Burrell, J.; Redlinger, C.; Gallagher, M.; Bradbury, A.R.; et al. Combination Paclitaxel and Palbociclib: Results of a Phase I Trial in Advanced Breast Cancer. Clin. Cancer Res. 2019, 25, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Vaughn, D.J.; Hwang, W.-T.; Lal, P.; Rosen, M.A.; Gallagher, M.; O’Dwyer, P.J. Phase 2 Trial of the Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Patients with Retinoblastoma Protein-Expressing Germ Cell Tumors. Cancer 2015, 121, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schönberger, S.; Calaminus, G.; Köhrer, K.; Albers, P.; et al. CDK4/6 Inhibition Presents as a Therapeutic Option for Paediatric and Adult Germ Cell Tumours and Induces Cell Cycle Arrest and Apoptosis via Canonical and Non-Canonical Mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR Inhibition Selectively Sensitizes G1 Checkpoint-Deficient Cells to Lethal Premature Chromatin Condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef]
- Bogani, G.; Lopez, S.; Mantiero, M.; Ducceschi, M.; Bosio, S.; Ruisi, S.; Sarpietro, G.; Guerrisi, R.; Brusadelli, C.; Dell’Acqua, A.; et al. Immunotherapy for Platinum-Resistant Ovarian Cancer. Gynecol. Oncol. 2020, 158, 484–488. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).