
Perspectives on Hypoxia Signaling in Tumor Stroma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Hypoxic Response of Tumor Vasculature
3. Hypoxic Response of ECM and CAFs
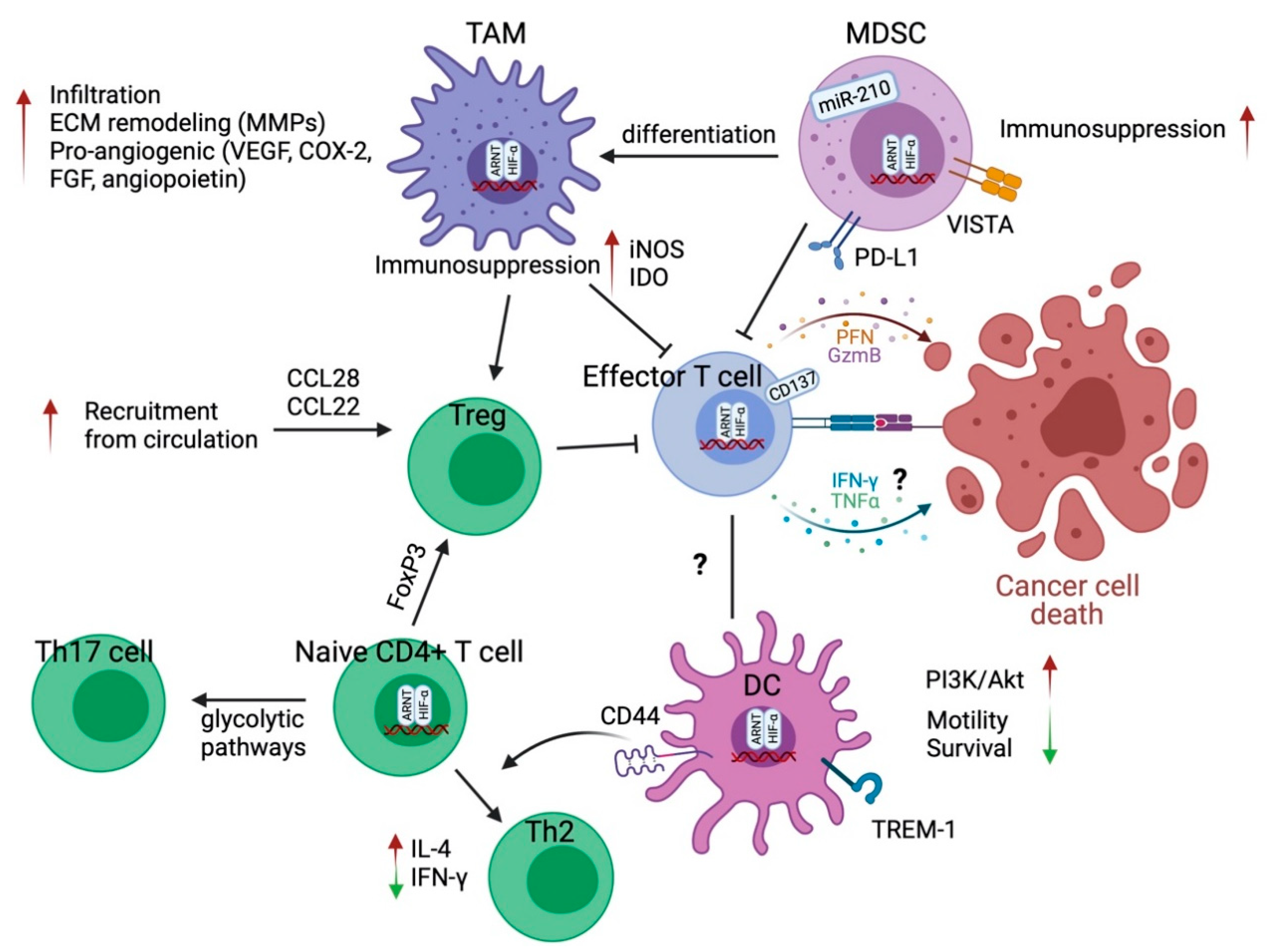
4. The Effect of Hypoxia on T Cells
4.1. CD4+ T Helper Cells and Regulatory T Cells
4.2. CD8+ Effector T Cells
5. The Effect of Hypoxia on Myeloid Cells
5.1. Tumor-Associated Macrophages
5.2. Myeloid-Derived Suppressor Cells (MDSCs)
5.3. Dendritic Cells
6. Targeting Hypoxia and HIFs in Cancer
6.1. Hypoxia Activated Prodrugs (HAPs)
6.2. Inhibitors of HIF
6.3. Combination with Immune Checkpoint Inhibitors
6.4. Combination with Anti-Angiogenesis Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Laitala, A.; Erler, J.T. Hypoxic signalling in tumour stroma. Front. Oncol. 2018, 8, 189. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Molecular Cell Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer. 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell. 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Welti, J.; Loges, S.; Dimmeler, S.; Carmeliet, P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Investig. 2013, 123, 3190–3200. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, K.Y.; Rivera, L.B.; Hur, H.; Carbon, J.G.; Toombs, J.E.; Goldstein, C.D.; Dellinger, M.T.; Castrillon, D.H.; Brekken, R.A. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014, 74, 1032–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenik, B.K.; Ostapoff, K.T.; Gerber, D.E.; Brekken, R.A. BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol. Cancer Ther. 2013, 12, 992–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, Y.; Reiberger, T.; Duyverman, A.M.; Huang, P.; Samuel, R.; Hiddingh, L.; Roberge, S.; Koppel, C.; Lauwers, G.Y.; et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology 2014, 59, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, N.N.; Kedrin, D.; Incio, J.; Liu, H.; Ho, W.W.; Nia, H.T.; Edrich, C.M.; Jung, K.; Daubriac, J.; Chen, I.; et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci. Transl. Med. 2016, 8, 360ra135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1 α and HIF2 α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Gunda, V.; Kumar, S.; Dasgupta, A.; Singh, P.K. Hypoxia-induced metabolomic alterations in pancreatic cancer cells. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1742, pp. 95–105. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dabrowski, M.; Collawn, J.F. Primary endothelial-specific regulation of hypoxiainducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial cell HIF-1α and HIF-2α differentially regulate metastatic success. Cancer Cell. 2012, 21, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Kuchnio, A.; Moens, S.; Bruning, U.; Kuchnio, K.; Cruys, B.; Thienpont, B.; Broux, M.; Ungureanu, A.A.; de Oliveira, R.L.; Bruyère, F.; et al. The cancer cell oxygen sensor PHD2 promotes metastasis via activation of cancer-associated fibroblasts. Cell Rep. 2015, 12, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Wright, S.; Zhang, J.; Brekken, R.A. Getting a grip on adhesion: Cadherin switching and collagen signaling. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118472. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Brekken, R.A. The next wave of stroma-targeting therapy in pancreatic cancer. Cancer Res. 2019, 79, 328–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. DMM Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Wang, Z.; Zhang, Y.; Brekken, R. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Myllyharju, J.; Schipani, E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010, 339, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Goggins, E.; Kakkad, S.; Mironchik, Y.; Jacob, D.; Wildes, F.; Krishnamachary, B.; Bhujwalla, Z.M. Hypoxia Inducible Factors Modify Collagen I Fibers in MDA-MB-231 Triple Negative Breast Cancer Xenografts. Neoplasia 2018, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Bajpai, S.; Wong, C.C.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Cell Death and Survival Procollagen Lysyl Hydroxylase 2 Is Essential for Hypoxia-Induced Breast Cancer Metastasis. Mol. Cancer Res. 2013, 11, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myllyharju, J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 2008, 40, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible f0actor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [Green Version]
- Sada, M.; Ohuchida, K.; Horioka, K.; Okumura, T.; Moriyama, T.; Miyasaka, Y.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; Nakamura, M. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett. 2016, 372, 210–218. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saatci, O.; Kaymak, A.; Raza, U.; Ersan, P.G.; Akbulut, O.; Banister, C.E.; Sikirzhytski, V.; Tokat, U.M.; Aykut, G.; Ansari, S.A.; et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Chiavarina, B.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Tanowitz, H.B.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Metabolic reprogramming and two-compartment tumor metabolism: Opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle 2012, 11, 3280–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Atkinson, S.J.; Neilson, L.J.; Lilla, S.; Knight, J.R.; Serneels, J.; Juin, A.; Ismail, S.; Bryant, D.M.; Markert, E.K.; et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci. Signal. 2019, 12, eaan8247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Evans, C.; Weidemann, A.; Takeda, N.; Lee, Y.S.; Stockmann, C.; Branco-Price, C.; Brandberg, F.; Leone, G.; Ostrowski, M.C.; et al. Loss of fibroblast HIF-1α accelerates tumorigenesis. Cancer Res. 2012, 72, 3187–3195. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.D.; Pedersen, J.T.; Venning, F.A.; Singh, L.B.; Moeendarbary, E.; Charras, G.; Cox, T.R.; Sahai, E.; Erler, J.T. Hypoxia and loss of PHD 2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015, 16, 1394–1408. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, T.; Kierstead, L.S.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Finke, J.H.; Bukowski, R.M.; Mueller-Berghaus, J.; Kirkwood, J.M.; Kwok, W.W.; et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB1*0401+ patients with renal cell carcinoma or melanoma. J. Exp. Med. 2002, 196, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Mattes, J.; Hulett, M.; Xie, W.; Hogan, S.; Rothenberg, M.E.; Foster, P.; Parish, C. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: An eotaxin and STAT6-dependent process. J. Exp. Med. 2003, 197, 387–393. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Makino, Y.; Okamoto, K.; Poellinger, L.; Ohnuma, K.; Morimoto, C.; Tanaka, H. TCR Engagement Increases Hypoxia-Inducible Factor-1α Protein Synthesis via Rapamycin-Sensitive Pathway under Hypoxic Conditions in Human Peripheral T Cells. J. Immunol. 2005, 174, 7592–7599. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehade, H.; Acolty, V.; Moser, M.; Oldenhove, G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J. Immunol. 2015, 195, 1372–1376. [Google Scholar] [CrossRef] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Darce, J.; Rudra, D.; Li, L.; Nishio, J.; Cipolletta, D.; Rudensky, A.Y.; Mathis, D.; Benoist, C. An N-Terminal Mutation of the Foxp3 Transcription Factor Alleviates Arthritis but Exacerbates Diabetes. Immunity 2012, 36, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.C. E3Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+ CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1α. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia enhances immunosuppression by inhibiting CD4+ Effector T cell function and promoting treg activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.S.; Lin, Y.L.; Wang, Y.A.; Mo, S.T.; Chi, P.Y.; Lai, A.C.; Pan, H.Y.; Chang, Y.J.; Lai, M.Z. HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8 + T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Palazón, A.; Martínez-Forero, I.; Teijeira, A.; Morales-Kastresana, A.; Alfaro, C.; Sanmamed, M.F.; Perez-Gracia, J.L.; Peñuelas, I.; Hervás-Stubbs, S.; Rouzaut, A.; et al. The HIF-1α hypoxia response in tumor-infi ltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov. 2012, 2, 608–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential Effects of Physiologically Relevant Hypoxic Conditions on T Lymphocyte Development and Effector Functions. J. Immunol. 2001, 167, 6140–6149. [Google Scholar] [CrossRef]
- de Almeida, P.E.; Mak, J.; Hernandez, G.; Jesudason, R.; Herault, A.; Javinal, V.; Borneo, J.; Kim, J.M.; Walsh, K.B. Anti-VEGF Treatment Enhances CD8+ T-Cell Antitumor Activity by Amplifying Hypoxia. Cancer Immunol. Res. 2020, 8, 806–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gropper, Y.; Feferman, T.; Shalit, T.; Salame, T.M.; Porat, Z.; Shakhar, G. Culturing CTLs under Hypoxic Conditions Enhances Their Cytolysis and Improves Their Anti-tumor Function. Cell Rep. 2017, 20, 2547–2555. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell. 2017, 32, 669–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, M.; Caldwell, C.C.; Kreth, S.; Kuboki, S.; Chen, P.; Smith, P.; Ohta, A.; Lentsch, A.B.; Lukashev, D.; Sitkovsky, M.V. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One 2007, 2, e853. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, D.; Klebanov, B.; Kojima, H.; Grinberg, A.; Ohta, A.; Berenfeld, L.; Wenger, R.H.; Ohta, A.; Sitkovsky, M. Cutting Edge: Hypoxia-Inducible Factor 1α and Its Activation-Inducible Short Isoform I.1 Negatively Regulate Functions of CD4 + and CD8 + T Lymphocytes. J. Immunol. 2006, 177, 4962–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.N.; Yang, J.F.; Huang, D.J.; Ni, H.H.; Zhang, C.X.; Zhang, L.; He, J.; Gu, J.M.; Chen, H.X.; Mai, H.Q.; et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11, 1906. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 2019, 4, e124989. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Henze, A.-T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of Macrophage Function during Tumor Progression: Regulation by Distinct Molecular Mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; VandenBerg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1α Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell. 2008, 13, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding Macrophage Entry into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.Y.; Chen, W.; Bai, X.L.; Xu, X.Y.; Zhang, Q.; Xia, X.F.; Sun, X.; Li, G.G.; Hu, Q.D.; Fu, Q.H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830. Available online: http://cancerres.aacrjournals.org/content/76/4/818.long (accessed on 1 August 2017). [CrossRef] [Green Version]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243. [Google Scholar] [CrossRef]
- White, J.R.; Harris, R.A.; Lee, S.R.; Craigon, M.H.; Binley, K.; Price, T.; Beard, G.L.; Mundy, C.R.; Naylor, S. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics 2004, 83, 1–8. [Google Scholar] [CrossRef]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowitzki, M.J.; Ritter, A.S.; Radhakrishnan, P.; Harnoss, J.M.; Opitz, V.M.; Biller, M.; Wehrmann, J.; Keppler, U.; Scheer, J.; Wallwiener, M.; et al. Pharmacological HIF-inhibition attenuates postoperative adhesion formation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Kimmer, G.; Wehrmann, J.; Ritter, A.S.; Radhakrishnan, P.; Opitz, V.M.; Tuffs, C.; Biller, M.; Kugler, J.; Keppler, U.; et al. Inhibition of HIF-prolyl hydroxylases improves healing of intestinal anastomoses. JCI Insight 2021, 6, e139191. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced: MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Hu, S.; Wu, J.C.; Martelli, F.; Bronte, V.; Chouaib, S. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. 2015, 75, 3771–3787. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Li, J.; Sarde, A.; Lines, J.L.; Lee, Y.C.; Qian, D.C.; Pechenick, D.A.; Manivanh, R.; Le Mercier, I.; Lowrey, C.H.; et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol. Res. 2019, 7, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Zhang, Q.; Shao, N.; Shan, Z.; Cheang, T.; Zhang, Z.; Su, Q.; Wang, S.; Lin, Y. Respiratory hyperoxia reverses immunosuppression by regulating myeloid-derived suppressor cells and PD-L1 expression in a triple-negative breast cancer mouse model. Am. J. Cancer Res. 2019, 9, 529–545. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30949408 (accessed on 4 February 2021). [PubMed]
- Tan, J.K.H.; O’Neill, H.C. Maturation requirements for dendritic cells in T cell stimulation leading to tolerance versus immunity. J. Leukoc. Biol. 2005, 78, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Bellone, G.; Carbone, A.; Smirne, C.; Scirelli, T.; Buffolino, A.; Novarino, A.; Stacchini, A.; Bertetto, O.; Palestro, G.; Sorio, C.; et al. Cooperative Induction of a Tolerogenic Dendritic Cell Phenotype by Cytokines Secreted by Pancreatic Carcinoma Cells. J. Immunol. 2006, 177, 3448–3460. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-β-secreting cells inducing CD4 +CD25 + regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Filippi, I.; Morena, E.; Aldinucci, C.; Carraro, F.; Sozzani, S.; Naldini, A. Short-Term hypoxia enhances the migratory capability of dendritic cell through HIF-1α and PI3K/Akt pathway. J. Cell Physiol. 2014, 229, 2067–2076. [Google Scholar] [CrossRef]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and Hypoxia-Inducible Factor-1α Modulate Lipopolysaccharide-Induced Dendritic Cell Activation and Function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Bosco, M.C.; Pierobon, D.; Blengio, F.; Raggi, F.; Vanni, C.; Gattorno, M.; Eva, A.; Novelli, F.; Cappello, P.; Giovarelli, M.; et al. Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: Identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 2011, 117, 2625–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierobon, D.; Bosco, M.C.; Blengio, F.; Raggi, F.; Eva, A.; Filippi, M.; Musso, T.; Novelli, F.; Cappello, P.; Varesio, L.; et al. Chronic hypoxia reprograms human immature dendritic cells by inducing a proinflammatory phenotype and TREM-1 expression. Eur. J. Immunol. 2013, 43, 949–966. [Google Scholar] [CrossRef]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting Edge: Inflammatory Responses Can Be Triggered by TREM-1, a Novel Receptor Expressed on Neutrophils and Monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef]
- Ogino, T.; Onishi, H.; Suzuki, H.; Morisaki, T.; Tanaka, M.; Katano, M. Inclusive estimation of complex antigen presentation functions of monocyte-derived dendritic cells differentiated under normoxia and hypoxia conditions. Cancer Immunol. Immunother. 2012, 61, 409–424. [Google Scholar] [CrossRef]
- Naldini, A.; Morena, E.; Pucci, A.; Miglietta, D.; Riboldi, E.; Sozzani, S.; Carraro, F. Hypoxia affects dendritic cell survival: Role of the hypoxia-inducible factor-1α and lipopolysaccharide. J. Cell. Physiol. 2012, 227, 587–595. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Ren, G.; Shao, Q.; Gao, W.; Sun, J.; Wang, H.; Ji, C.; Li, X.; Zhang, Y.; et al. Increased expression of surface CD44 in hypoxia-DCs skews helper T cells toward a Th2 polarization. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.W.; Gold, M.J.; Garcia-Batres, C.; Tai, K.; Elford, A.R.; Himmel, M.E.; Elia, A.J.; Ohashi, P.S. Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity. PLoS ONE 2020, 15, e0244366. [Google Scholar] [CrossRef] [PubMed]
- Kheshtchin, N.; Arab, S.; Ajami, M.; Mirzaei, R.; Ashourpour, M.; Mousavi, N.; Khosravianfar, N.; Jadidi-Niaragh, F.; Namdar, A.; Noorbakhsh, F.; et al. Inhibition of HIF-1α enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol. Immunother. 2016, 65, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xie, Y.; Xu, S.; Xin, J.; Wang, J.; Han, T.; Ting, R.; Zhang, J.; An, F. Hypoxia-activated nanomedicines for effective cancer therapy. Eur. J. Med. Chem. 2020, 195, 112274. [Google Scholar] [CrossRef]
- Doherty, N.; Hancock, S.L.; Kaye, S.; Coleman, C.N.; Shulman, L.; Marquez, C.; Mariscal, C.; Rampling, R.; Senan, S.; Roemeling, R.V. Muscle cramping in phase i clinical trials of tirapazamine (SR 4233) with and without radiation. Int. J. Radiat. Oncol. 1994, 29, 379–382. [Google Scholar] [CrossRef]
- Baran, N.; Konopleva, M. Molecular pathways: Hypoxia-activated prodrugs in cancer therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [Green Version]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Liapis, V.; Labrinidis, A.; Zinonos, I.; Hay, S.; Ponomarev, V.; Panagopoulos, V.; DeNichilo, M.; Ingman, W.; Atkins, G.J.; Findlay, D.M.; et al. Hypoxia-activated pro-drug TH-302 exhibits potent tumor suppressive activity and cooperates with chemotherapy against osteosarcoma. Cancer Lett. 2015, 357, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sun, J.D.; Wang, J.; Ahluwalia, D.; Baker, A.F.; Cranmer, L.D.; Ferraro, D.; Wang, Y.; Duan, J.X.; Ammons, W.S.; et al. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: Optimization of dosing regimens and schedules. Cancer Chemother. Pharmacol. 2012, 69, 1487–1498. [Google Scholar] [CrossRef] [Green Version]
- Lohse, I.; Rasowski, J.; Cao, P.; Pintilie, M.; Do, T.; Tsao, M.S.; Hill, R.P.; Hedley, D.W. Targeting hypoxic microenvironment of pancreatic xenografts with the hypoxia-activated prodrug TH-302. Oncotarget 2016, 7, 33571–33580. [Google Scholar] [CrossRef] [Green Version]
- Saggar, J.K.; Tannock, I.F. Chemotherapy Rescues Hypoxic Tumor Cells and Induces Their Reoxygenation and Repopulation—An Effect That Is Inhibited by the Hypoxia-Activated Prodrug TH-302. Clin. Cancer Res. 2015, 21, 2107–2114. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.G.; Zegers, C.M.; Biemans, R.; Lieuwes, N.G.; van Stiphout, R.G.; Yaromina, A.; Sun, J.D.; Hart, C.P.; Windhorst, A.D.; van Elmpt, W.; et al. TH-302 in Combination with Radiotherapy Enhances the Therapeutic Outcome and Is Associated with Pretreatment [18 F]HX4 Hypoxia PET Imaging. Clin. Cancer Res. 2015, 21, 2984–2992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Marrano, P.; Wu, B.; Kumar, S.; Thorner, P.; Baruchel, S. Combined Antitumor Therapy with Metronomic Topotecan and Hypoxia-Activated Prodrug, Evofosfamide, in Neuroblastoma and Rhabdomyosarcoma Preclinical Models. Clin. Cancer Res. 2016, 22, 2697–2708. [Google Scholar] [CrossRef] [Green Version]
- Codony, V.L.; Tavassoli, M. Hypoxia-induced therapy resistance: Available hypoxia-targeting strategies and current advances in head and neck cancer. Transl. Oncol. 2021, 14, 101017. [Google Scholar] [CrossRef]
- Hunter, F.W.; Wouters, B.G.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personalised medicine. Br. J. Cancer. 2016, 114, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Eliasof, S.; Lazarus, D.; Peters, C.G.; Case, R.I.; Cole, R.O.; Hwang, J.; Schluep, T.; Chao, J.; Lin, J.; Yen, Y.; et al. Correlating preclinical animal studies and human clinical trials of a multifunctional, polymeric nanoparticle. Proc. Natl. Acad. Sci. USA 2013, 110, 15127–15132. [Google Scholar] [CrossRef] [Green Version]
- Pham, E.; Yin, M.; Peters, C.G.; Lee, C.R.; Brown, D.; Xu, P.; Man, S.; Jayaraman, L.; Rohde, E.; Chow, A.; et al. Preclinical Efficacy of Bevacizumab with CRLX101, an Investigational Nanoparticle–Drug Conjugate, in Treatment of Metastatic Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 4493–4503. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Lin, Y.-L.; Luh, F.; Yen, Y.; Chen, R.-M. Preclinical effects of CRLX101, an investigational camptothecin-containing nanoparticle drug conjugate, on treating glioblastoma multiforme via apoptosis and antiangiogenesis. Oncotarget 2016, 7, 42408–42421. [Google Scholar] [CrossRef] [Green Version]
- Schluep, T.; Hwang, J.; Cheng, J.; Heidel, J.D.; Bartlett, D.W.; Hollister, B.; Davis, M.E. Preclinical Efficacy of the Camptothecin-Polymer Conjugate IT-101 in Multiple Cancer Models. Clin. Cancer Res. 2006, 12, 1606–1614. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Li, Z.; Zink, J.I.; Tamanoi, F. In vivo tumor suppression efficacy of mesoporous silica nanoparticles-based drug-delivery system: Enhanced efficacy by folate modification. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.T.; Chau, C.H.; Strope, J.D.; Huitema, A.D.; Sissung, T.M.; Price, D.K.; Figg, W.D. Antitumor activity of NLG207 (formerly CRLX101) in combination with enzalutamide in preclinical prostate cancer models. Mol. Cancer Ther. 2021, 20, 915–924. [Google Scholar] [CrossRef]
- Krasner, C.N.; Birrer, M.J.; Berlin, S.T.; Horowitz, N.S.; Buss, M.K.; Eliasof, S.; Garmey, E.G.; Hennessy, M.G.; Konstantinopoulos, P.; Matulonis, U. Phase II clinical trial evaluating CRLX101 in recurrent ovarian, tubal, and peritoneal cancer. J. Clin. Oncol. 2014, 32, 5581. [Google Scholar] [CrossRef]
- Xie, C.; Gao, X.; Sun, D.; Zhang, Y.; Krausz, K.W.; Qin, X.; Gonzalez, F.J. Metabolic profiling of the novel hypoxia-inducible factor 2α inhibitor PT2385 in vivo and in vitro. Drug Metab. Dispos. 2018, 46, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Strowd, R.E.; Ellingson, B.M.; Wen, P.Y.; Ahluwalia, M.S.; Piotrowski, A.F.; Desai, A.S.; Clarke, J.L.; Lieberman, F.S.; Desideri, S.; Nabors, L.B.; et al. Safety and activity of a first-in-class oral HIF2-alpha inhibitor, PT2385, in patients with first recurrent glioblastoma (GBM). J. Clin. Oncol. 2019, 37, 2027. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible Factor 1α Is Regulated by the Mammalian Target of Rapamycin (mTOR) via an mTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [Green Version]
- Bohan, P.M.; Chick, R.C.; O’Shea, A.E.; Vreeland, T.J.; Hickerson, A.T.; Cindass, J.L.; Ensley, D.C.; Hale, D.; Clifton, G.T.; Sohn, V.Y.; et al. Phase I Trial of Encapsulated Rapamycin in Patients with Prostate Cancer Under Active Surveillance to Prevent Progression. Cancer Prev. Res. 2021, 14, 551–562. [Google Scholar] [CrossRef]
- Ugwueze, C.V.; Ogamba, O.J.; Young, E.E.; Onyenekwe, B.M.; Ezeokpo, B.C. Metformin: A Possible Option in Cancer Chemotherapy. Anal. Cell. Pathol. 2020, 2020, 7180923. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Saluja, A. Minnelide, a novel drug for pancreatic and liver cancer. Pancreatology 2015, 15, S39–S43. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Mowery, Y.M.; Mitchell, J.B.; Cherukuri, M.K.; Secomb, T.W. Rationale for hypoxia assessment and amelioration for precision therapy and immunotherapy studies. J. Clin. Investig. 2019, 129, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Melillo, G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat. Rev. Clin. Oncol. 2012, 9, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Cao, Z.; Zhou, Q. Progress in tumor vascular normalization for anticancer therapy: Challenges and perspectives. Front. Med. 2012, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Lee, H.J.; Park, D.J.; Lee, Y.J.; Tap, W.D.; Eisinger-Mathason, T.S.; Hart, C.P.; Choy, E.; Simon, M.C.; Yoon, S.S. Hypoxia-activated chemotherapeutic TH-302 enhances the effects of VEGF-A inhibition and radiation on sarcomas. Br. J. Cancer 2015, 113, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Wang, T.; Chen, X.; Yang, R.; Kumar, S.M.; Vultur, A.; Li, P.; Martin, J.S.; Herlyn, M.; et al. Hypoxia-activated prodrug enhances therapeutic effect of sunitinib in melanoma. Oncotarget 2017, 8, 115140. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sun, J.D.; Zhang, L.; Mokhtari, R.B.; Wu, B.; Meng, F.; Liu, Q.; Bhupathi, D.; Wang, Y.; Yeger, H.; et al. Hypoxia-Targeting Drug Evofosfamide (TH-302) Enhances Sunitinib Activity in Neuroblastoma Xenograft Models. Transl. Oncol. 2018, 11, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Floyd, J.; Fichtel, L.; Michalek, J.; Kanakia, K.P.; Huang, S.; Reardon, D.; Wen, P.Y.; Lee, E.Q. Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci. Rep. 2021, 11, 2306. [Google Scholar] [CrossRef] [PubMed]
- Meaney, C.; Rhebergen, S.; Kohandel, M. In silico analysis of hypoxia activated prodrugs in combination with anti angiogenic therapy through nanocell delivery. PLoS Comput. Biol. 2020, 16, e1007926. [Google Scholar] [CrossRef] [PubMed]
- Fjeldbo, C.S.; Hompland, T.; Hillestad, T.; Aarnes, E.K.; Günther, C.C.; Kristensen, G.B.; Malinen, E.; Lyng, H. Combining imaging- and gene-based hypoxia biomarkers in cervical cancer improves prediction of chemoradiotherapy failure independent of intratumour heterogeneity. EbioMedicine 2020, 57, 102841. [Google Scholar] [CrossRef] [PubMed]
- Bernauer, C.; Man, Y.K.S.; Chisholm, J.C.; Lepicard, E.Y.; Robinson, S.P.; Shipley, J.M. Hypoxia and its therapeutic possibilities in paediatric cancers. Br. J. Cancer. 2021, 124, 539–551. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Ritter, A.S.; Kimmer, G.; Schneider, M. Hypoxia-adaptive pathways: A pharmacological target in fibrotic disease? Pharmacol. Res. 2019, 147, 104364. [Google Scholar] [CrossRef]
- Haase, V.H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. 2017, 21, S110–S124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, T.W.; Sternlicht, M.D.; Klaus, S.J.; Neff, T.B.; Liu, D.Y. Induction of erythropoiesis by hypoxia-inducible factor prolyl hydroxylase inhibitors without promotion of tumor initiation, progression, or metastasis in a VEGF-sensitive model of spontaneous breast cancer. Hypoxia 2017, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Clinical Trial | Trial Phase | Drug | Mechanism | Disease | References |
|---|---|---|---|---|---|
| Hypoxia activated prodrug | |||||
| NCT03224182 | III | Apaziquone | Indolequinone | Non-muscle invasive bladder cancer | Clinicaltrials.gov |
| NCT02174549 | I/II | Tirapazamine + transarterial embolization | Aromatic n-oxide | Liver cancer | Clinicaltrials.gov |
| NCT01880359 NCT02661152 | III | Nimorazole + chemoradiotherapy | 5-nitroimidazoles, radiosensitizer | Locally advanced head and neck squamous cell cancer | Clinicaltrials.gov |
| HIF inhibitors | |||||
| NCT03531827 | II | NLG207 + enzalutamide | HIF-1α/topoisomerase I inhibitor + antiandrogen | Metastatic prostate cancer | [158] |
| NCT02769962 NCT04669002 | I/II | EP0057 + olaparib | HIF-1α/topoisomerase I inhibitor + PARP inhibitor | Relapsed/refractory small cell lung cancer Ovarian cancer | Clinicaltrials.gov |
| NCT03108066 NCT03216499 | II | PT2385 | HIF-2α inhibitor | Von Hippel-Lindau associated clear cell renal carcinoma Glioblastoma | Clinicaltrials.gov |
| NCT02974738 | I | Belzutifan (PT2977) | HIF-2α inhibitor | Advanced solid tumors | Clinicaltrials.gov |
| NCT04195750 | III | Belzutifan (MK-6482) | HIF-2α inhibitor | Advanced renal cell carcinoma | Clinicaltrials.gov |
| Targeting Associated pathways | |||||
| NCT01101438 NCT01864096 NCT01697566 NCT03685409 | III | Metformin | Decreases HIF-1α accumulation | Early-stage breast cancer Low risk prostate cancer Chemoprevention study in endometrial and oral cancer | [149] |
| NCT02614339 | III | Metformin + traditional chemotherapy | Decreases HIF-1α accumulation | Recurrent colorectal cancer | [149] |
| NCT04275713 | II | Metformin + cisplatin | Decreases HIF-1α accumulation | Locally advanced cervical cancer | [179] Clinicaltrials.gov |
| NCT03117920 | II | Minnelide | HSP70, p300 inhibitors | Refractory pancreatic cancer | [149] |
| NCT03450018 | I/II | SLC0111+ gemcitabine | CAIX | Metastatic pancreatic cancer | [180] |
| NCT04648033 NCT02628080 | I | Atovaquone + chemoradiotherapy | Antimalarial drug; Hypoxia modifier via inhibition of mitochondrial complex III | Locally advanced non-small cell lung cancer | [180] Clinicaltrials.gov |
| Combination with immunotherapy or anti-angiogenesis therapy | |||||
| NCT03098160 | I | TH-302 + ipilimumab | HAP + anti-CTLA4 Ab | Advanced solid malignancies | [170] |
| NCT01652079 | II | NLG207 + bevacizumab | HIF-1α/topoisomerase I inhibitor + anti-VEGF Ab | Ovarian/peritoneal cancer | [158] |
| NCT03634540 | II | Belzutifan (PT2977) + cabozantinib | HIF-2α + VEGFR2 inhibitors | Advanced clear cell renal carcinoma | Clinicaltrials.gov |
| NCT04895748 | I/Ib | DFF332 + everolimus + spartalizumab | HIF-2α inhibitor + mTOR inhibitor + anti-PD-1 Ab | Relapsed renal cell carcinoma, advanced malignancies with HIF stabilizing mutations | Clinicaltrials.gov |
| NCT04114136 | II | Metformin or rosiglitazone + nivolumab or pembrolizumab | Decreases HIF-1α accumulation | Advanced solid tumor malignancies | Clinicaltrials.gov |
| Assessment of hypoxia | |||||
| NCT03003637 | IB/II | 18F-FDG PET-CT | Pre and post nivolumab +/- ipilimumab | Advanced/recurrent head and neck carcinoma | [171] |
| NCT03373994 | 18F-FDG PET-CT | Evaluate tumor perfusion and hypoxia | Solid tumors | Clinicaltrials.gov | |
| NCT03646747 | Oxygen enhanced MRI measurement | Pre and post radiotherapy | Head and neck cancer | Clinicaltrials.gov | |
| NCT04309552 | FMISO, FLT PET | Compare FMISO, FLT PET vs. molecular biomarkers of hypoxia and cell proliferation | High grade glioma | Clinicaltrials.gov | |
| NCT02095249 | Pimonidazole followed by prostatectomy | Measure tumor hypoxia via immunohistochemical staining | Prostate cancer | Clinicaltrials.gov | |
| NCT04001023 | 18F-EF5 PET-CT and targeted tumor sampling | Identify molecular differences between hypoxic and non-hypoxic tumors | Advanced ovarian cancer | Clinicaltrials.gov | |
| NCT00568490 | Osteopontin, lysyl oxidase, macrophage inhibiting factor and proteomic technology | Identify hypoxic biomarkers in blood and tumors | Head and neck cancer Lung cancer | Clinicaltrials.gov | |
| NCT03054792 | 18F-FAZA/BOLD PET-MRI | Measure hypoxia between start and completion of treatment | Pediatric sarcomas | Clinicaltrials.gov | |
| Hypoxia assessment + radiotherapy | |||||
| NCT04846309 | I | FMISO PET + radiation | Hypoxic tumors receive higher dose of radiation | Esophageal cancer | Clinicaltrials.gov |
| NCT02352792 | II | FMISO PET + radiation | Hypoxic tumors receive 10% higher dose of radiation | Head and neck squamous cell carcinoma | Clinicaltrials.gov |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Coleman, M.; Brekken, R.A. Perspectives on Hypoxia Signaling in Tumor Stroma. Cancers 2021, 13, 3070. https://doi.org/10.3390/cancers13123070
Zhang Y, Coleman M, Brekken RA. Perspectives on Hypoxia Signaling in Tumor Stroma. Cancers. 2021; 13(12):3070. https://doi.org/10.3390/cancers13123070
Chicago/Turabian StyleZhang, Yuqing, Morgan Coleman, and Rolf A. Brekken. 2021. "Perspectives on Hypoxia Signaling in Tumor Stroma" Cancers 13, no. 12: 3070. https://doi.org/10.3390/cancers13123070