
Ca2+ Signaling and Its Potential Targeting in Pancreatic Ductal Carcinoma

 , , , , ,
, , , , , 
 ,
,  , ,
, ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Ca2+ Transporters in PDAC
2.1. Channels
2.2. Pumps
2.3. Transporters
3. Role of Ca2+ Signaling in PDAC Pathophysiology
3.1. PDAC Cells and Autophagy
3.2. PDAC Cells and Proliferation
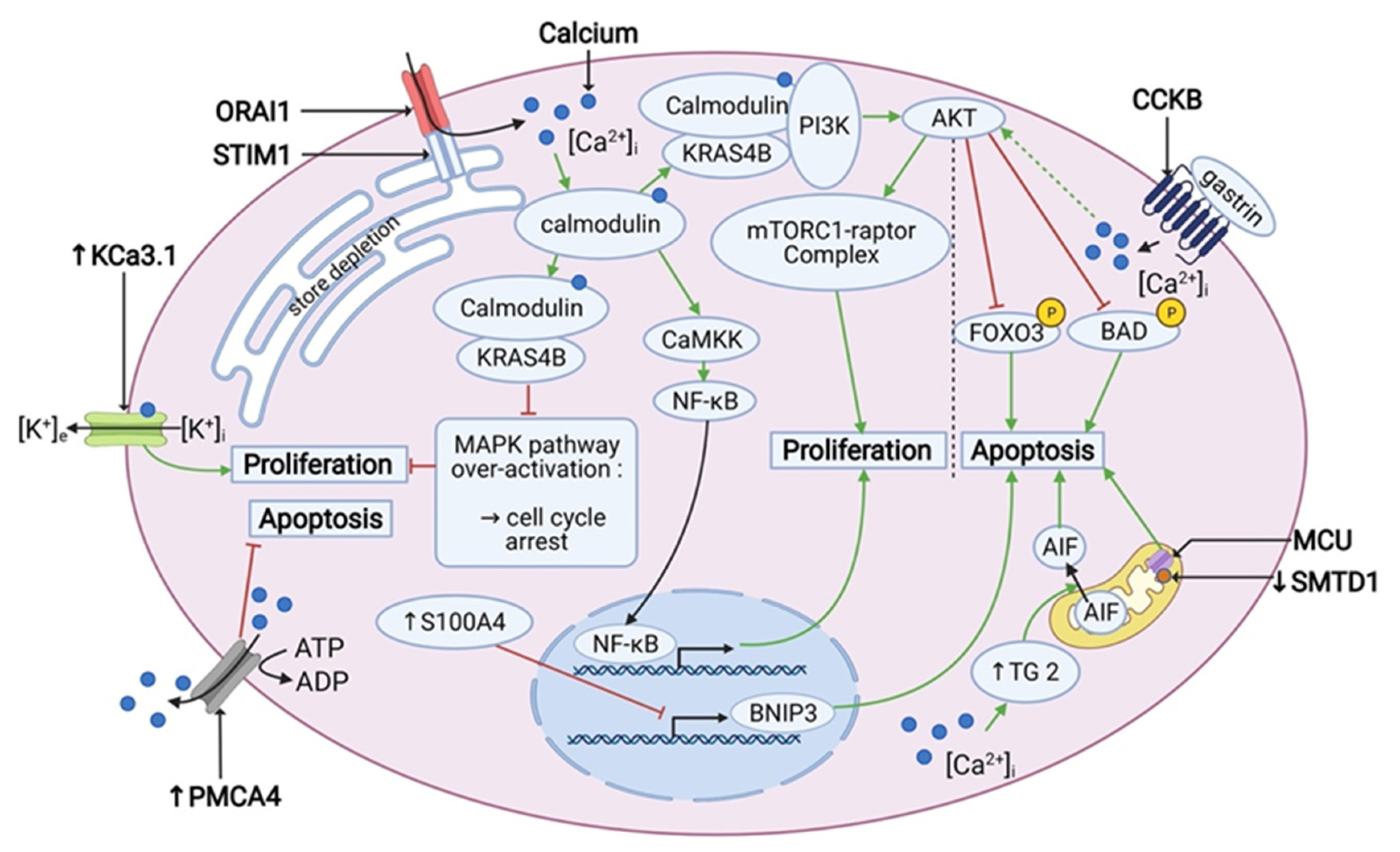
3.3. K-RAS, Calmodulin and Proliferation
3.4. PDAC Cells and Apoptosis
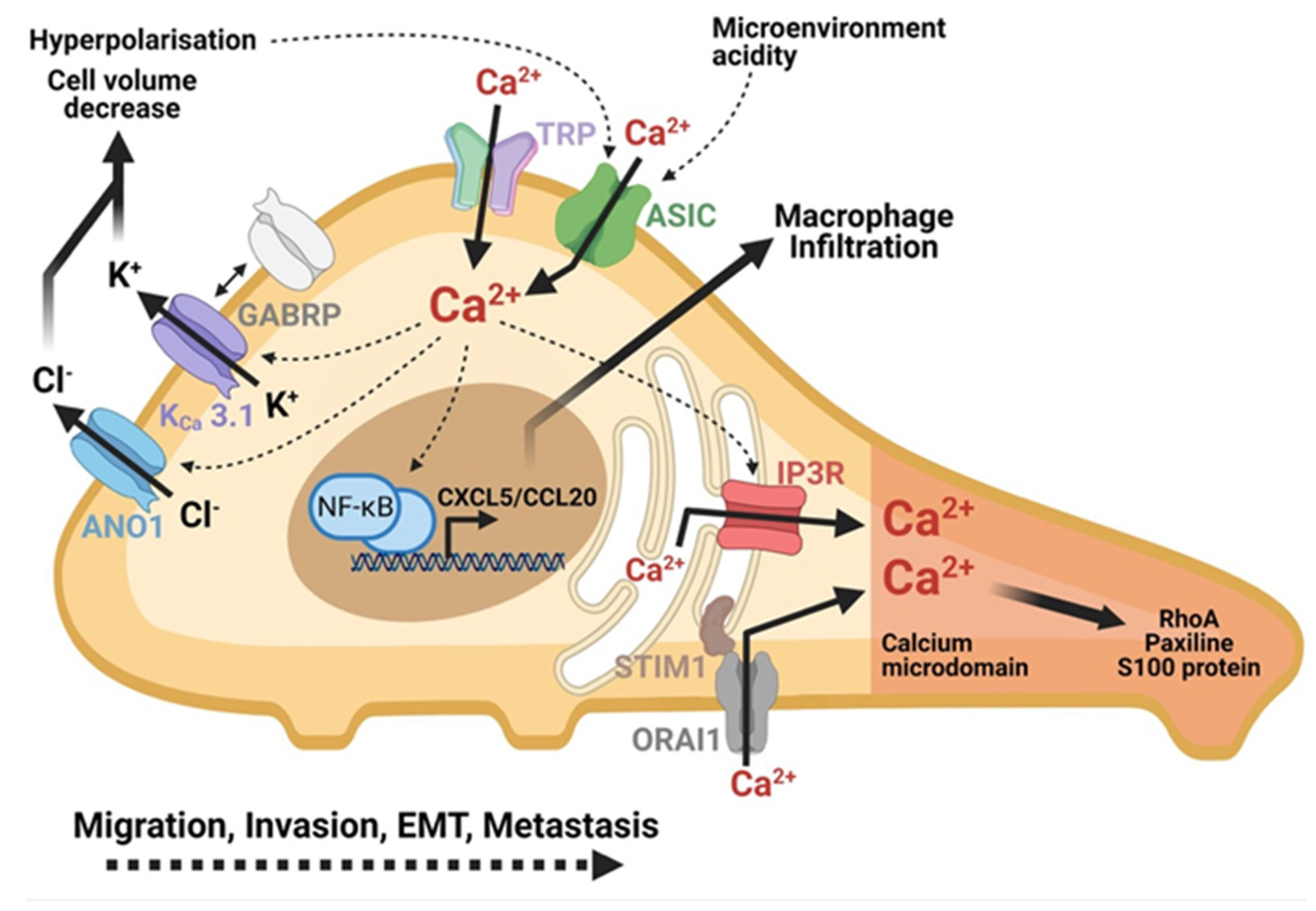
3.5. Role of Ca2+ Signaling in PDAC Cell Motility
3.6. Role of Ca2+ Signaling in PDAC Cell Invasion
3.7. Role of Ca2+ Signaling in PDAC Metastasis Formation
4. Ca2+ Signaling in PDAC Treatment
4.1. Are Ca2+ Transporters Potential Targets for PDAC Treatment?
4.1.1. Impact on Proliferation
4.1.2. Impact on Migration and Invasion
4.1.3. Impact on Some Other Mechanisms
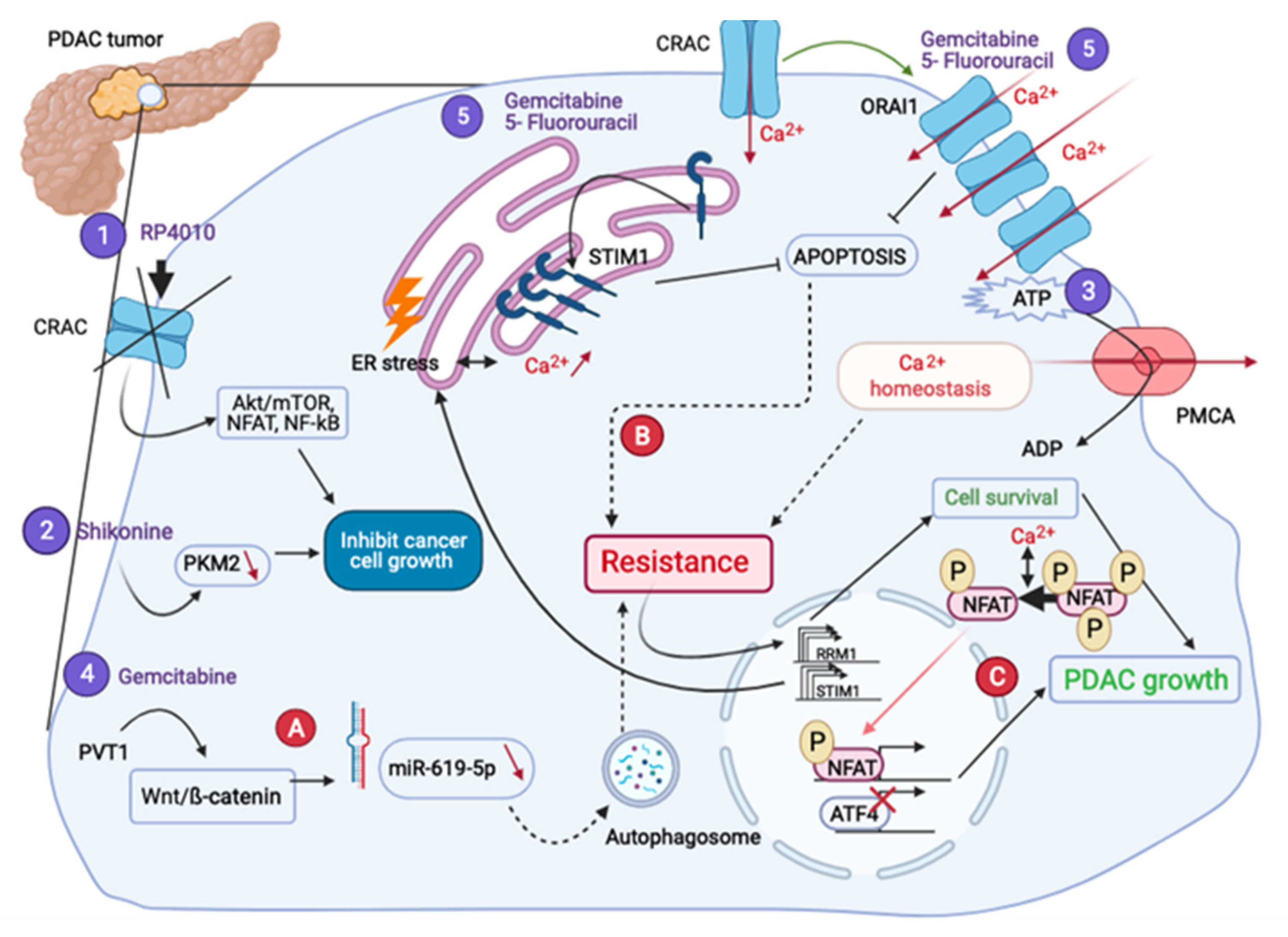
4.2. What Is Their Place in Drug Resistance?
5. General Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Institut National Du Cancer-Accueil. Available online: https://www.e-cancer.fr/ (accessed on 14 April 2021).
- Ansari, D.; Tingstedt, B.; Andersson, B.; Holmquist, F.; Sturesson, C.; Williamsson, C.; Sasor, A.; Borg, D.; Bauden, M.; Andersson, R. Pancreatic Cancer: Yesterday, Today and Tomorrow. Future Oncol. 2016, 12, 1929–1946. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 12 April 2021).
- Ohuchida, K.; Mizumoto, K.; Miyasaka, Y.; Yu, J.; Cui, L.; Yamaguchi, H.; Toma, H.; Takahata, S.; Sato, N.; Nagai, E.; et al. Over-Expression of S100A2 in Pancreatic Cancer Correlates with Progression and Poor Prognosis. J. Pathol. 2007, 213, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic Cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of Pancreatic Cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Rao, R. Calcium-ATPases: Gene Disorders and Dysregulation in Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1344–1350. [Google Scholar] [CrossRef]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.-M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient Receptor Potential Melastatin-Related 7 Channel Is Overexpressed in Human Pancreatic Ductal Adenocarcinomas and Regulates Human Pancreatic Cancer Cell Migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef] [PubMed]
- Storck, H.; Hild, B.; Schimmelpfennig, S.; Sargin, S.; Nielsen, N.; Zaccagnino, A.; Budde, T.; Novak, I.; Kalthoff, H.; Schwab, A. Ion channels in control of pancreatic stellate cell migration. Oncotarget 2016, 8, 769–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fels, B.; Nielsen, N.; Schwab, A. Role of TRPC1 channels in pressure-mediated activation of murine pancreatic stellate cells. Eur. Biophys. J. 2016, 45, 657–670. [Google Scholar] [CrossRef]
- Okeke, E.; Parker, T.; Dingsdale, H.; Concannon, M.; Awais, M.; Voronina, S.G.; Molgó, J.; Begg, M.; Metcalf, D.; Knight, A.E.; et al. Epithelial–mesenchymal transition, IP3 receptors and ER–PM junctions: Translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 2016, 473, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.-F.; Guo, D.-D.; Luo, X.-J. SMDT1-driven change in mitochondrial dynamics mediate cell apoptosis in PDAC. Biochem. Biophys. Res. Commun. 2019, 511, 323–329. [Google Scholar] [CrossRef] [PubMed]
- James, A.D.; Patel, W.; Butt, Z.; Adiamah, M.; Dakhel, R.; Latif, A.; Uggenti, C.; Swanton, E.; Imamura, H.; Siriwardena, A.K.; et al. The Plasma Membrane Calcium Pump in Pancreatic Cancer Cells Exhibiting the Warburg Effect Relies on Glycolytic ATP. J. Biol. Chem. 2015, 290, 24760–24771. [Google Scholar] [CrossRef] [Green Version]
- Kendrick, A.A.; Schafer, J.; Dzieciatkowska, M.; Nemkov, T.; D’Alessandro, A.; Neelakantan, D.; Ford, H.L.; Pearson, C.G.; Weekes, C.D.; Hansen, K.C.; et al. CD147: A small molecule transporter ancillary protein at the crossroad of multiple hallmarks of cancer and metabolic reprogramming. Oncotarget 2016, 8, 6742–6762. [Google Scholar] [CrossRef] [Green Version]
- Sritangos, P.; Alarcon, E.P.; James, A.D.; Sultan, A.; Richardson, D.A.; Bruce, J.I.E. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 2020, 12, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.S.; Mandal, C.; Albiez, R.S.; Samanta, S.K.; Mandal, C. Mahanine drives pancreatic adenocarcinoma cells into endoplasmic reticular stress-mediated apoptosis through modulating sialylation process and Ca2+-signaling. Sci. Rep. 2018, 8, 3911. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shao, Z.; Chen, S.; Shi, L.; Li, Z. A SLC24A2 Gene Variant Uncovered in Pancreatic Ductal Adenocarcinoma by Whole Exome Sequencing. Tohoku J. Exp. Med. 2017, 241, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Gouaux, E. Principles of Selective Ion Transport in Channels and Pumps. Science 2005, 310, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E.; Runnels, L.; Strübing, C. The trp ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hong, C.; Kim, B.J.; So, I. The Pathophysiologic Roles of TRPM7 Channel. Korean J. Physiol. Pharmacol. 2014, 18, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Prokaryotic Cytoskelet. 2018, 87, 141–165. [Google Scholar] [CrossRef]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but Differential Localization and Recruitment of STIM1, Orai1 and TRPC Channels in Secretory Cells. Traffic 2010, 12, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- James, A.D.; Richardson, D.A.; Oh, I.-W.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I.E. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: Role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 2020, 122, 266–278. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2010, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Baba, A.; Matsuda, T.; Djamgoz, M.B.; Yaqoob, M.M.; Eccles, S.A. Ca2+ Influx through Reverse Mode Na+/Ca2+ Exchange Is Critical for Vascular Endothelial Growth Factor-mediated Extracellular Signal-regulated Kinase (ERK) 1/2 Activation and Angiogenic Functions of Human Endothelial Cells*. J. Biol. Chem. 2011, 286, 37919–37931. [Google Scholar] [CrossRef] [Green Version]
- Caspersen, C.; Pedersen, P.S.; Treiman, M. The Sarco/Endoplasmic Reticulum Calcium-ATPase 2b Is an Endoplasmic Reticulum Stress-inducible Protein. J. Biol. Chem. 2000, 275, 22363–22372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, A.H.; Frandsen, A.; Treiman, M. Upregulation of the SERCA-type Ca2+ pump activity in response to endoplasmic reticulum stress in PC12 cells. BMC Biochem. 2001, 2, 4. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D. The mitochondrial calcium uniporter is a highly selective ion channel. Nat. Cell Biol. 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 Regulates Orai1 Trafficking and Store Independent Ca2+ Entry in a Model of Lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nat. Cell Biol. 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 8048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Padmanabhan, J.; Chellappan, S. The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression. Cancers 2015, 7, 1447–1471. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.-D.; Xia, X.; Lv, X.-F.; Yu, B.-X.; Yuan, J.-N.; Mai, X.-Y.; Shang, J.-Y.; Zhou, J.-G.; Liang, S.-J.; Pang, R.-P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2016, 21, 904–915. [Google Scholar] [CrossRef]
- Berry, C.T.; May, M.J.; Freedman, B.D. STIM- and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium 2018, 74, 131–143. [Google Scholar] [CrossRef]
- Klee, C.B.; Vanaman, T.C. Calmodulin. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1982; Volume 281, pp. 213–321. [Google Scholar]
- Agell, N.; Aliguévicençalemany, R.; Castro, A.; Jaime, M.; Pujol, M.J.; Rius, E.; Serratosa, J.; Taulés, M.; Bachs, O. New nuclear functions for calmodulin. Cell Calcium 1998, 23, 115–121. [Google Scholar] [CrossRef]
- Grant, R.C.; Denroche, R.; Jang, G.H.; Nowak, K.M.; Zhang, A.; Borgida, A.; Holter, S.; Topham, J.T.; Wilson, J.; Dodd, A.; et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut 2020. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Wrzeszczynski, K.O.; Rahman, S.; Frank, M.O.; Arora, K.; Shah, M.; Geiger, H.; Felice, V.; Manaa, D.; Dikoglu, E.; Khaira, D.; et al. Identification of targetable BRAF ΔN486_P490 variant by whole-genome sequencing leading to dabrafenib-induced remission of a BRAF-mutant pancreatic adenocarcinoma. Mol. Case Stud. 2019, 5, a004424. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.Y.; Marzullo, G.; Hine, B. Calmodulin plays a pivotal role in cellular regulation. Science 1980, 207, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Bachs, O.; Agell, N.; Carafoli, E. Calmodulin and calmodulin-binding proteins in the nucleus. Cell Calcium 1994, 16, 289–296. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Villalonga, P.; López-Alcalá, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N. Calmodulin Binds to K-Ras, but Not to H- or N-Ras, and Modulates Its Downstream Signaling. Mol. Cell. Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Planchon, S.M.; Wolfman, J.C.; Wolfman, A. Growth Factor-dependent AKT Activation and Cell Migration Requires the Function of c-K(B)-Ras Versus Other Cellular Ras Isoforms. J. Biol. Chem. 2006, 281, 29730–29738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, S.J.; Nolet, R.P.; Calvert, R.J.; Anderson, L.M.; Gaponenko, V. The Hypervariable Region of K-Ras4B Is Responsible for Its Specific Interactions with Calmodulin. Biochemistry 2009, 48, 7575–7583. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Wolfman, J.C.; Wolfman, A. K-Ras Regulates the Steady-state Expression of Matrix Metalloproteinase 2 in Fibroblasts. J. Biol. Chem. 2003, 278, 31871–31878. [Google Scholar] [CrossRef] [Green Version]
- Bonito, B.; Sauter, D.R.P.; Schwab, A.; Djamgoz, M.B.A.; Novak, I. KCa3.1 (IK) modulates pancreatic cancer cell migration, invasion and proliferation: Anomalous effects on TRAM-34. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1865–1875. [Google Scholar] [CrossRef]
- Jäger, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of Intermediate-Conductance Ca2+-Activated K+ Channels Inhibit Human Pancreatic Cancer Cell Growth in Vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Gao, W.; Li, X.; Yu, L.; Luo, D.; Liu, Y.; Yu, X. S100A14 promotes progression and gemcitabine resistance in pancreatic cancer. Pancreatology 2021. [Google Scholar] [CrossRef]
- Mahon, P.C.; Baril, P.; Bhakta, V.; Chelala, C.; Caulee, K.; Harada, T.; Lemoine, N.R. S100A4 Contributes to the Suppression of BNIP3 Expression, Chemoresistance, and Inhibition of Apoptosis in Pancreatic Cancer. Cancer Res. 2007, 67, 6786–6795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.A.; Sritangos, P.; James, A.D.; Sultan, A.; Bruce, J.I.E. Metabolic regulation of calcium pumps in pancreatic cancer: Role of phosphofructokinase-fructose-bisphosphatase-3 (PFKFB3). Cancer Metab. 2020, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fok, J.Y.; Mehta, K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis 2007, 12, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Stepan, V.; Todisco, A. Intracellular mechanisms mediating the anti-apoptotic action of gastrin. Biochem. Biophys. Res. Commun. 2004, 323, 44–48. [Google Scholar] [CrossRef]
- Fels, B.; Bulk, E.; Pethő, Z.; Schwab, A. The Role of TRP Channels in the Metastatic Cascade. Pharmaceuticals 2018, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Wang, Y.; Chen, Q.; Liu, Z.; Xiao, S.; Wang, B.; Shi, B. TRPM2 promotes the proliferation and invasion of pancreatic ductal adenocarcinoma. Mol. Med. Rep. 2018, 17, 7537–7544. [Google Scholar] [CrossRef]
- Song, H.; Dong, M.; Zhou, J.; Sheng, W.; Li, X.; Gao, W. Expression and prognostic significance of TRPV6 in the development and progression of pancreatic cancer. Oncol. Rep. 2018, 39, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nat. Cell Biol. 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, H.-Y.; Deng, S.-C.; He, C.; Li, X.; Chen, J.-Y.; Jin, Y.; Hu, Z.-L.; Wang, F.; Wang, C.-Y.; et al. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Novak, I. Molecular basis of potassium channels in pancreatic duct epithelial cells. Channels 2013, 7, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Sauter, D.R.P.; Novak, I.; Pedersen, S.F.; Larsen, E.H.; Hoffmann, E.K. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflügers Arch. Eur. J. Physiol. 2015, 467, 1495–1508. [Google Scholar] [CrossRef] [Green Version]
- Crottès, D.; Lin, Y.-H.T.; Peters, C.; Gilchrist, J.M.; Wiita, A.P.; Jan, Y.N.; Jan, L.Y. TMEM16A controls EGF-induced calcium signaling implicated in pancreatic cancer prognosis. Proc. Natl. Acad. Sci. USA 2019, 116, 13026–13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.Y.; Mpilla, G.B.; Sexton, R.; Viswanadha, S.; Penmetsa, K.V.; Aboukameel, A.; Diab, M.; Kamgar, M.; Al-Hallak, M.N.; Szlaczky, M.; et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers 2020, 12, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, W.; Junling, S.; Kaili, Z.; Jinmeng, H.; Jiuxing, D.; Jianwei, S.; Wang, J.; Shen, J.; Zhao, K.; Hu, J.; et al. STIM1 overexpression in hypoxia microenvironment contributes to pancreatic carcinoma progression. Cancer Biol. Med. 2019, 16, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. ZEB1 and IL-6/11-STAT3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of S100 proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Liang, L.; Luo, R.; Ding, Y.; Liu, K.; Shen, L.; Zeng, H.; Ge, Y.; Zeng, M. S100A4 overexpression in pancreatic ductal adenocarcinoma: Imaging biomarkers from whole-tumor evaluation with MRI and texture analysis. Abdom. Radiol. 2021, 46, 623–635. [Google Scholar] [CrossRef]
- Jiang, S.-H.; Zhu, L.-L.; Zhang, M.; Li, R.-K.; Yang, Q.; Yan, J.-Y.; Zhang, C.; Dong, F.-Y.; Dai, M.; Hu, L.-P.; et al. GABRP regulates chemokine signalling, macrophage recruitment and tumour progression in pancreatic cancer through tuning KCNN4-mediated Ca2+ signalling in a GABA-independent manner. Gut 2019, 68, 1994–2006. [Google Scholar] [CrossRef]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural Regulation of Hematopoiesis, Inflammation, and Cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef] [Green Version]
- Demir, I.E.; Friess, H.; Ceyhan, G.O. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front. Physiol. 2012, 3, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Ishizuka, J.; Cooper, C.W.; Chung, D.H.; Tsuchiya, T.; Uchida, T.; Rajaraman, S.; Townsend, C.M.; Thompson, J.C. Inhibitory Effect of Calcium Channel Blockers on Growth of Pancreatic Cancer Cells. Pancreas 1994, 9, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Alhothali, M.; Mathew, M.; Iyer, G.; Lawrence, H.R.; Yang, S.; Chellappan, S.; Padmanabhan, J. Fendiline Enhances the Cytotoxic Effects of Therapeutic Agents on PDAC Cells by Inhibiting Tumor-Promoting Signaling Events: A Potential Strategy to Combat PDAC. Int. J. Mol. Sci. 2019, 20, 2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passacantilli, I.; Panzeri, V.; Terracciano, F.; Fave, G.D.; Sette, C.; Capurso, G. Co-treatment with gemcitabine and nab-paclitaxel exerts additive effects on pancreatic cancer cell death. Oncol. Rep. 2018, 39, 1984–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadros, S.B.; Shukla, S.K.; King, R.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Lyudmyla, B.; Lazenby, A.J.; Berim, L.; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Yi, C.; Yi, Y.; Qin, W.; Yan, Y.; Dong, X.; Zhang, X.; Huang, Y.; Zhang, R.; Wei, J.; et al. LncRNA PVT1 promotes gemcitabine resistance of pancreatic cancer via activating Wnt/β-catenin and autophagy pathway through modulating the miR-619-5p/Pygo2 and miR-619-5p/ATG14 axes. Mol. Cancer 2020, 19, 1–24. [Google Scholar] [CrossRef]
- Kutschat, A.P.; Hamdan, F.H.; Wang, X.; Wixom, A.Q.; Najafova, Z.; Gibhardt, C.S.; Kopp, W.; Gaedcke, J.; Ströbel, P.; Ellenrieder, V.; et al. STIM1 Mediates Calcium-Dependent Epigenetic Reprogramming in Pancreatic Cancer. Cancer Res. 2021, 81, 2943–2955. [Google Scholar] [CrossRef]
- James, A.D.; Chan, A.K.; Erice, O.; Siriwardena, A.K.; Bruce, J.I.E. Glycolytic ATP Fuels the Plasma Membrane Calcium Pump Critical for Pancreatic Cancer Cell Survival. J. Biol. Chem. 2013, 288, 36007–36019. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Gu, L.; Li, H.; Gao, Y.; Li, X.; Shen, D.; Gong, H.; Li, S.; Niu, S.; Zhang, Y.; et al. Hypoxia-induced overexpression of stanniocalcin-1 is associated with the metastasis of early stage clear cell renal cell carcinoma. J. Transl. Med. 2015, 13, 56. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.-H.; Li, T.; Gong, Y.-Q.; He, Q.; Shen, Q.; Peng, S.-H.; Wang, J.-Y.; Chen, J.-C.; Guo, Y.-L.; Gong, K. Association between FBP1 and hypoxia-related gene expression in clear cell renal cell carcinoma. Oncol. Lett. 2016, 11, 4095–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Wang, N. Upregulation of ASPM, BUB1B and SPDL1 in tumor tissues predicts poor survival in patients with pancreatic ductal adenocarcinoma. Oncol. Lett. 2020, 19, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunda, V.; Kumar, S.; Dasgupta, A.; Singh, P.K. Hypoxia-Induced Metabolomic Alterations in Pancreatic Cancer Cells. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; pp. 95–105. [Google Scholar]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Dejos, C.; Gkika, D.; Cantelmo, A.R. The Two-Way Relationship between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol. 2020, 8, 573747. [Google Scholar] [CrossRef] [PubMed]
- Krška, Z.; Svab, J.; Hoskovec, D.; Ulrych, J. Pancreatic Cancer Diagnostics and Treatment—Current State. Prague Med. Rep. 2015, 116, 253–267. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calcium Transporters [RT1] | Calcium Transporter Type | Localization | Alteration | Involved Cancers | Ref |
|---|---|---|---|---|---|
| Channels | |||||
| TRPM7 (transient receptor potential melastatin related 7) | Cationic (Ca2+) channels | Plasma membrane | Migration and/or invasion of cancer cells: metastasis formation | PDACPancreatic ductal adeno carcinoma, epidermal, neuroblastoma, glioblastoma, breast cancer, nasopharynx cancer, lung cancer, prostate cancer | [10] |
| TRPC3 (Transient Receptor Potential Cation Channel Subfamily C Member) | Cationic (Ca2+) channels | Plasma membrane | Proliferation and metastasis formation | Pancreatic ductal adenocarcinoma | [11] |
| TRPC1 (Transient Receptor Potential Cation Channel Subfamily C Member) | Mechanosensitive channel | Plasma membrane | Altered TRPC1 with tissue pressure | Pancreatic ductal adenocarcinoma | [12] |
| Orai1 (SOCC: store-operated Ca2+ Channel) | Calcium channel | Plasma membrane | Migration and/or invasion of cancer cells: metastasis formation | Pancreatic ductal adenocarcinoma | [13] |
| IP3R (Inositol trisphosphate receptor) | Receptor channel | Endoplasmic reticulum membrane | Migration and/or invasion of cancer cells: metastasis formation | Pancreatic ductal adenocarcinoma | [13] |
| Pumps & Transporters | |||||
| MCU (mitochondrial calcium uniporter complex) | Transmembrane receptor | Membrane of mitochondria | Altered mitochondrial dynamics | Pancreatic ductal adenocarcinoma | [14] |
| SMDT1 (Single-Pass Membrane Protein with Aspartate Rich Tail 1) | Essential regulatory subunit of the MCU | ||||
| PMCA (plasma membrane Ca2+ ATPase) | Transport protein of Calcium (pump) | Plasma membrane | High calcium efflux | Pancreatic ductal adenocarcinoma | [15] |
| PMCA1 (plasma membrane Ca2+ ATPase isoform 1) | Transport protein of Calcium | Plasma membrane | Upregulated | Pancreatic ductal adenocarcinoma | [16] |
| PMCA4 (plasma membrane Ca2+ ATPase isoform 4) | Transport protein of Calcium | Plasma membrane | Cell migration and apoptotic resistance | Pancreatic ductal adenocarcinoma | [17] |
| SERCA2 (sarco/endoplasmic reticulum Ca2+-ATPase) | Calcium ATPase-type Pump-ATPase | Endoplasmic reticulum membrane | Upregulated | Pancreatic ductal adenocarcinoma | [18] |
| SLC24A2 (NCX1,2,3) (Solute Carrier Family 24 Member 2) | Sodium/Potassium/Calcium Exchanger | Plasma membrane | Mutation in SLC24A2 gene decreases the stability of SLC24A2, tumor progression | Pancreatic ductal adenocarcinoma | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettaieb, L.; Brulé, M.; Chomy, A.; Diedro, M.; Fruit, M.; Happernegg, E.; Heni, L.; Horochowska, A.; Housseini, M.; Klouyovo, K.; et al. Ca2+ Signaling and Its Potential Targeting in Pancreatic Ductal Carcinoma. Cancers 2021, 13, 3085. https://doi.org/10.3390/cancers13123085
Bettaieb L, Brulé M, Chomy A, Diedro M, Fruit M, Happernegg E, Heni L, Horochowska A, Housseini M, Klouyovo K, et al. Ca2+ Signaling and Its Potential Targeting in Pancreatic Ductal Carcinoma. Cancers. 2021; 13(12):3085. https://doi.org/10.3390/cancers13123085
Chicago/Turabian StyleBettaieb, Louay, Maxime Brulé, Axel Chomy, Mel Diedro, Malory Fruit, Eloise Happernegg, Leila Heni, Anaïs Horochowska, Mahya Housseini, Kekely Klouyovo, and et al. 2021. "Ca2+ Signaling and Its Potential Targeting in Pancreatic Ductal Carcinoma" Cancers 13, no. 12: 3085. https://doi.org/10.3390/cancers13123085
APA StyleBettaieb, L., Brulé, M., Chomy, A., Diedro, M., Fruit, M., Happernegg, E., Heni, L., Horochowska, A., Housseini, M., Klouyovo, K., Laratte, A., Leroy, A., Lewandowski, P., Louvieaux, J., Moitié, A., Tellier, R., Titah, S., Vanauberg, D., Woesteland, F., ... Lehen’kyi, V. (2021). Ca2+ Signaling and Its Potential Targeting in Pancreatic Ductal Carcinoma. Cancers, 13(12), 3085. https://doi.org/10.3390/cancers13123085